


作者:田小幺
编辑:李宝珠
转载请联系本公众号获得授权,并标明来源
哈佛大学与博世公司的联合研究团队提出创新性解决方案,开发出电学响应的统一可微学习框架。该框架可在单一机器学习模型中同时学习广义势能及其对外部刺激的响应函数,通过将响应函数定义为广义势能对原子坐标与扰动参数的导数,利用两者间的精确数学关系,严格满足动量守恒、Born 电荷声学求和规则等物理约束,克服了传统独立模型的固有缺陷,为晶体、无序及液态材料介电和铁电性质的高精度研究开辟了新路径。
计算材料科学作为现代材料科学领域的前沿研究方向,承担着解析材料微观结构与预测宏观性能的关键使命。该学科以第一性原理为基石,基于量子力学等基础物理定律,致力于精确预测真实材料的可实验测量性质,从而深入理解材料在外部刺激下的响应机制。这些响应特性涵盖线性、非线性及耦合效应,是决定电介质、铁电体、多铁材料和压电材料等功能表现的核心要素。
当前,基于密度泛函理论(DFT)的第一性原理电子结构方法虽为研究材料性质的重要手段,但受限于计算成本随系统规模呈指数增长的特性,仅能处理小规模体系,极大制约了对复杂材料体系的系统性研究。近年来,机器学习方法的引入为这一领域带来突破,其通过构建数据驱动模型,在预测材料极化、Born 电荷、极化率、介电常数、光谱特性等方面展现出强大潜力,并已成功应用于分子、液态水及固体材料体系。
然而,现有多数机器学习方法仍存在局限性,例如独立设计的模型难以确保物理对称性和守恒定律的严格执行;部分尝试整合介电性质、能量与力计算的单模型方案,在扩展至具有周期性边界条件和极化多值性的实际体系时面临挑战。尽管有研究通过在不同电场下训练原子力构建势能面,间接推导极化特性,以规避多值极化数据训练难题,但该方法依赖隐式导数计算,可能降低预测精度,且大规模 DFT 数据采集的高成本问题仍未得到根本解决。
针对上述挑战,哈佛大学联合德国博世集团在美国的子公司 Robert Bosch LLC,共同开发出电学响应的统一可微学习框架。该框架可在单一机器学习模型中同时学习广义势能及其对外部刺激的响应函数,通过将响应函数定义为广义势能对原子坐标与扰动参数的导数,利用两者间的精确数学关系,严格满足动量守恒、Born 电荷声学求和规则等物理约束,克服了传统独立模型的固有缺陷,为晶体、无序及液态材料介电和铁电性质的高精度研究开辟了新路径。
相关研究成果以「Unified differentiable learning of electric response」为题,已发表于国际权威期刊 Nature Communications。
研究亮点:
* 首创统一机器学习框架,基于广义势能和可观测响应量之间的精确微分关系,实现动量、电焓等多维度守恒保障。
* 开发等变神经网络模型,突破百万原子级铁电滞回模拟瓶颈,精准解析极化切换的畴成核与一维扩展动力学。
* 解决极化多值性训练难题,结合第一性原理在 α−SiO₂/BaTiO₃ 体系中实现介电与铁电性质的跨尺度高精度预测。

论文地址:
https://go.hyper.ai/18TWg
关注公众号,后台回复「电学响应」获取完整 PDF
更多 AI 前沿论文:
https://go.hyper.ai/owxf6
α−SiO₂ 与 BaTiO₃ 数据实验,训练集构建与关键参数推导
该研究针对 α−SiO₂ 和 BaTiO₃ 两种材料开展数据实验,通过构建训练集与验证集实现模型性能验证。
在训练数据生成阶段,α−SiO₂ 的 200 帧原子构型从 300 K 和 600 K 的 NVT 经典 MD 模拟中提取(Vashishta 势驱动,各运行 100 ps,1 ps 间隔采样)。BaTiO₃ 则通过 FLARE 代码的主动学习动力学收集 75 帧(300 K–400 K,原始结构 60 帧与畴壁结构 15 帧)。
每组数据均通过 DFT 计算无电场时的能量、力和极化,并在 0.36 MV/cm 的小电场下利用有限差分法推导 Born 电荷和极化率,确保极化与电场的线性响应范围。
基于等变神经网络的材料响应预测框架
在本研究中,研究人员成功开发出一种创新性的机器学习框架,致力于实现对广义势能与响应函数的统一学习。该框架严格遵循泰勒展开的数学原理,巧妙地在一个统一的模型架构内,同步开展广义势能及其导数的学习任务,从而精准预测材料的响应特性。
这一模型建构的核心理念在于,将广义势能关于其关键变量(例如原子位置以及外部场)进行求导操作,由此能够为每一个原子构型自动化地生成与之对应的响应特性,涵盖力、极化以及 Born 电荷等诸多方面。如此设计,不仅确保了物理对称性以及诸如动量守恒、电焓守恒等守恒定律得以精准贯彻,而且还显著提高了模型的预测精度和可靠性。
在模型的训练环节,研究人员采用的方式是通过将描述系统扰动的参数添加到输入中,使得模型能够对这些参数进行微分,从而在额外的物理量上进行训练。同时,研究人员还最小化一个全面整合了能量、力、极化以及 Born 电荷等多种响应特性贡献因素的综合性损失函数。这种训练模式与 Sobolev 训练的概念存在着紧密的关联性,具体而言,组成损失函数的各个项,均由训练标签与能量对应的梯度之间的差值构成。
为了精准捕捉响应特性对于多个场的非线性依赖关系以及耦合特性,研究人员选用了神经网络架构,并借助梯度反向传播这一强大技术手段,深入学习广义势能与其输入之间错综复杂的关系网络。
在模型架构的搭建过程中,研究人员依托 Allegro 平台,充分利用等变神经网络所具备的高精度优势以及出众的数据效率,同时,通过严格遵循局部性设计理念,实现了模型卓越的可拓展性。在模型的输入设计方面,如图所示,研究人员巧妙地将电场 E 与原子位置 r_i 进行融合,这使得模型得以同步开展对电焓 U、力 F_i、极化 P、Born 电荷 Z_i 以及极化率 α 的学习任务。
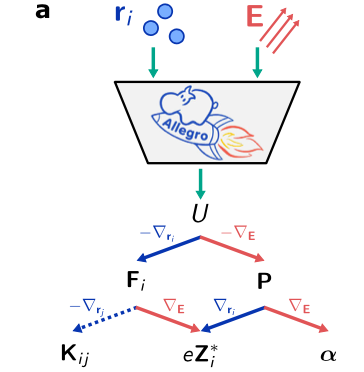
神经网络公式的示意图
在训练数据的生成过程中,如下图所示,研究人员在零电场周边区域,借助 DFT 计算进行数据获取,并运用有限差分近似方法来确定 Born 电荷以及极化率的相关数值。完成训练后,模型具备输出电焓的能力,进而通过对输出电焓进行一阶以及二阶导数的运算,推导出力、极化、Born 电荷以及极化率等一系列关键参数。
值得一提的是,研究人员专门开发了相应的接口,使得该模型能够与 LAMMPS 软件实现无缝集成,从而有力支持在电场环境条件下开展大规模结构弛豫以及机器学习分子动力学(Machine Learning Molecular Dynamics, MLMD)模拟工作。
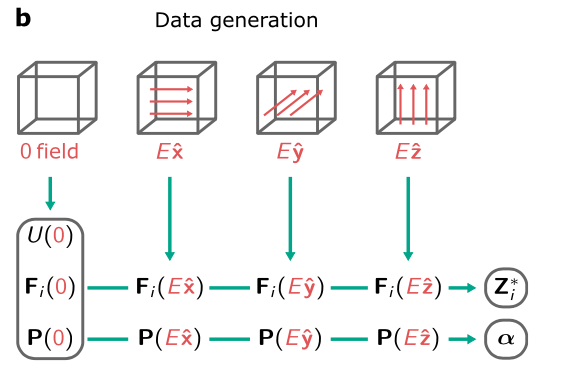
给定一组有限电场框架计算生成DFT数据的工作流程
模型准确性的多场景验证
为验证模型性能,该研究围绕材料振动与介电性质、铁电滞回及偶极动力学两大方向展开实验,以 α−SiO₂ 和 BaTiO₃ 为研究对象,深入探究模型在不同场景下的准确性与适用性。
在振动和介电性质研究中,研究人员选择 α−SiO₂ 作为典型研究对象,从经典势能的分子动力学模拟中提取了 72 个原子的 200 帧数据,并基于此训练出相应的机器学习模型。为深入探究模型的准确性,构建了含 24,696 个原子的超胞,在 NVE 系综下平衡 10 皮秒后,开展了 200 皮秒无电场的机器学习分子动力学(MLMD)模拟。通过极化动力学计算红外光谱,并分析极化和极化率动力学以确定频率依赖的介电常数,最终将结果与基于密度泛函微扰理论(DFPT)的计算结果及实验数据进行对比。
如下图 a-c 所示,MLMD 与 DFPT 的结果高度吻合。此外,在研究电场存在下电子和离子的屏蔽效应时,研究人员基于 α−SiO₂ 原始体相结构,在有限电场下进行结构弛豫。通过计算电场与无电场时极化差异确定静态介电常数,结果如下图 d 所示,模型所得的高频和静态介电常数与 DFT 值基本一致,这充分表明该模型能够精准捕捉电场对电子结构的贡献,验证了其在有限电场动力学模拟中的有效性。
同时,在 Born 电荷的训练验证中发现,若不在 Born 电荷上训练模型,低频下振动和介电响应的准确性会受到显著影响,尤其是在数据有限的情况下更为明显,并且会增加电场下模拟的计算成本。这一发现凸显了在 Born 电荷上训练是该模型的关键优势。
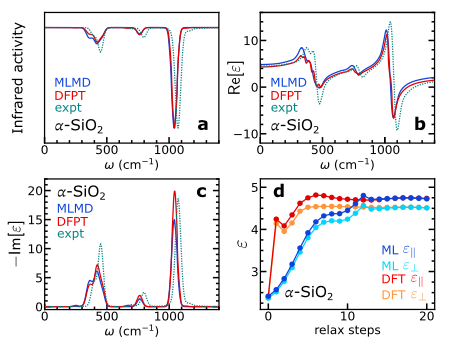
α−SiO₂ 的介电性质
在铁电滞回和偶极动力学研究中,研究人员聚焦于 BaTiO₃ 钙钛矿,采用主动学习动力学提取的 75 帧、每帧 135 个原子的数据训练机器学习模型。对 135 个原子超胞在零温度下计算铁电滞回,结果如下图 a 所示,MLMD 模拟与 DFT 计算所得铁电滞回高度一致,有力地验证了模型的可靠性。
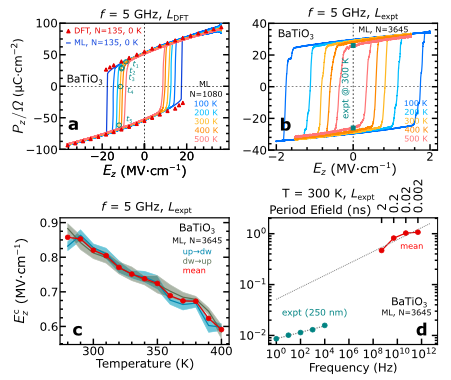
BaTiO₃ 的铁电性质
为进一步评估温度对铁电响应的影响,研究人员在 NVT 系综、5 GHz 正弦变化电场下开展 MLMD。研究发现,如上图 b-c 所示,随着温度升高,固有矫顽场减小,而自发极化受温度影响相对较小。无论是在零温度还是有限温度下,滞回曲线均呈现出电场符号对称性,这符合极化保守矢量场的特性。此外,使用实验晶格参数的 3,645 个原子超胞进行研究时,自发极化与实验结果相符,矫顽场也更接近实验值,同时验证了模型在不同原子数和晶格参数超胞下的外推能力。
在探究电场频率对铁电滞回的影响时,3,645 个原子超胞在 300 K 下的 MLMD 显示,矫顽场随频率降低而减小。尽管外推后与实验值仍存在约一个数量级的差异,但这一复杂计算任务凸显了模型强大的计算能力以及对百万原子级系统的出色扩展性能。
材料模型研究:学术与企业界的协同共进
在材料科学领域,学术界与企业界纷纷发力,多点开花,共同推动着材料模型相关研究的持续发展。
对于学术界而言,众多高校与科研团队成果斐然。美国罗切斯特大学的科研人员开发出一款机器学习模型,能够分析 X 射线衍射(XRD)实验产生的海量数据,加速材料创新。该模型经不同实验条件和晶体特性的无机材料实验数据训练,并依据布拉格定律分类,优化架构。英国帝国理工学院提出的 Chemeleon 模型,借助生成式 AI 依据材料结构特性数据集导航,从文本描述和三维结构数据中学习,实现化学成分和晶体结构的采样。韩国首尔大学与美国福特汉姆大学合作团队运用大型语言模型(LLM)预测新材料可合成性,并阐释预测依据,展现出良好性能和可解释性。中国东南大学物理学院王金兰、马亮教授团队联合南京大学王欣然教授团队,在二维材料研究方面提出新思路,实现厘米级均匀双层 MoS₂ 薄膜的层数可控外延生长,并通过计算模拟提出提升接触界面性能的方法,成功实现超低接触电阻。
企业界也不甘示弱,积极投身材料模型技术的创新与应用。苹果公司运用机器学习模型优化金属合金配方,用于产品外壳制造,提升材料强度与耐用性,降低成本。巴斯夫开发先进材料模型,模拟材料性能,加速新型塑料与涂料研发,提高产品竞争力。微软推出的 MatterGen 模型,通过独特扩散模型架构生成符合设计要求的材料结构,相比传统方法优势显著,并与中国科学院 SIAT 团队合作得到新型材料 TaCr₂O₆。
这些前沿探索与创新实践相互交织,持续推动材料模型研究向纵深发展。未来,随着研究不断深入与技术迭代升级,材料模型将在更广领域实现突破与应用,为科技发展筑牢根基。
参考文章:
1.https://mp.weixin.qq.com/s/mctu0DOWO_OieLnOgp93Rw
2.https://mp.weixin.qq.com/s/I-UZTyUFSWwXlf1LCmwjRQ
3.https://mp.weixin.qq.com/s/Ox62ut3IJcUWsLC7sF100Q
4.https://mp.weixin.qq.com/s/VlPb8zSghVVxnPNl-WzqBA
5.https://plastics-rubber.basf.com/global/en/performance_polymers/services/service_ultrasim/Material-Modeling



戳“阅读原文”,免费获取海量数据集资源!
(文:HyperAI超神经)