


作者:哇塞
编辑:李宝珠
转载请联系本公众号获得授权,并标明来源
麻省理工学院和哈佛大学的团队提出了一种结合了蛋白质序列和细胞图像来进行未知蛋白质亚细胞定位的预测框架 PUPS。该框架创新地结合了蛋白质语言模型和图像修复模型来预测蛋白质定位,使其兼并推向未知蛋白预测的泛化能力和捕获细胞可变性的细胞类型特定预测。
蛋白质亚细胞定位(subcellular localization of a protein)是指蛋白质在细胞结构中具体的定位情况,这对蛋白质行使其生物学功能至关重要。举个简单例子,如果把细胞想象成一个庞大的企业,其中细胞核、线粒体、细胞膜等对应总裁办、发电部、门岗等不同的部门,那么只有对应的蛋白进入正确的「部门」才能使其正常工作,否则便会导致某些疾病发生,如癌症、阿尔兹海默病。因此,精准定位蛋白质亚细胞可以说是生命科学的核心任务之一。
尽管科研界已经对不同细胞系中的数千种蛋白质进行了空间定位分析,但到目前为止,已测量的蛋白质与细胞系组合数量还只是其中的冰山一角。比如当前最大的亚细胞定位数据集——人类蛋白质图谱(Human Protein Atlas,HPA),提供了 13,147 个基因编码的蛋白质亚细胞定位(占已知人类蛋白质编码基因的 65%),但是整个数据集包含了 37 个细胞系,而每种蛋白质最多只能在其中三株中进行测量。与此同时,主流的实验手段很难在同一细胞中同时检测所有蛋白质数量,这严重阻碍了全面分析复杂的蛋白质网络,增加了实验复杂度和误差风险。
除此之外,蛋白质定位并非静止不变的,它的变异性不仅体现在细胞系之间,甚至在同一细胞系的单个细胞间也会发生,而现有数据图谱记录的蛋白质和细胞系对仅反映了特定条件下的结果。因此,即便是现有成果也很难直接套用,需要根据环境变化而对蛋白质定位进一步探索。
为了解决蛋白质亚细胞定位技术方法的局限性和生物系统复杂性之间的矛盾,机器学习被寄予厚望。如今已经建模并成功应用的如基于蛋白质序列的模型、基于细胞图像的模型等,虽然在某些方面表现亮眼,但不足之处也十分突出——前者忽视了细胞类型的特异性定位差异,后者则缺乏推向未知蛋白研究的泛化能力。
有鉴于此,来自美国麻省理工学院和哈佛大学的研究团队提出了一种结合蛋白质序列和细胞图像来进行未知蛋白质亚细胞定位的预测框架,命名为 Predictions of Unseen Proteins’ Subcellular localization(PUPS)。PUPS 创新地结合了蛋白质语言模型和图像修复模型来预测蛋白质定位,使其兼并推向未知蛋白预测的泛化能力和捕获细胞可变性的细胞类型特定预测。实验证明,该框架能够准确预测训练数据集之外新实验中蛋白质的定位,具有极佳的泛化能力和高度的准确性,应用潜力突出。
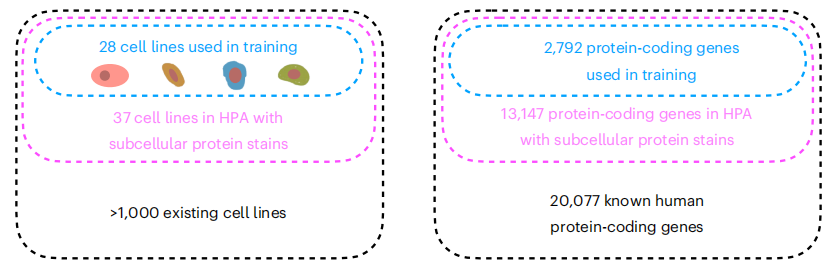
PUPS 技术研究背景,目标及现有数据的局限性
研究成果以「Prediction of protein subcellular localization in single cells」为题,已发表于 Nature Methods。
研究亮点:
* 所提研究创新地结合了蛋白质语言模型和图像绘制模型,利用蛋白质序列和细胞图像进行蛋白质定位预测,弥补了过往计算模型的不足
* PUPS 能够推广到未知蛋白质和细胞系,从而评估细胞系之间以及细胞系内单个细胞间蛋白质定位的变异性,并识别与具有可变定位的蛋白质相关的生物过程
* 在训练数据集之外的新实验中,PUPS 同样展示了其高度精确的预测能力,具有突出的应用潜力和医学价值

论文地址:
https://go.hyper.ai/LeaQF
关注公众号,后台回复「蛋白质定位」获取完整 PDF
数据集地址:
数据集:以尽可能全面的数据打造可信模型
PUPS 的训练数据集来自于人类蛋白质图谱(Human Protein Atlas,HPA),研究团队将第 16 版 HPA 数据汇总到第 22 版当中,以尽可能多的收集蛋白质的数据,确保实验分析的全面性。如下图所示:
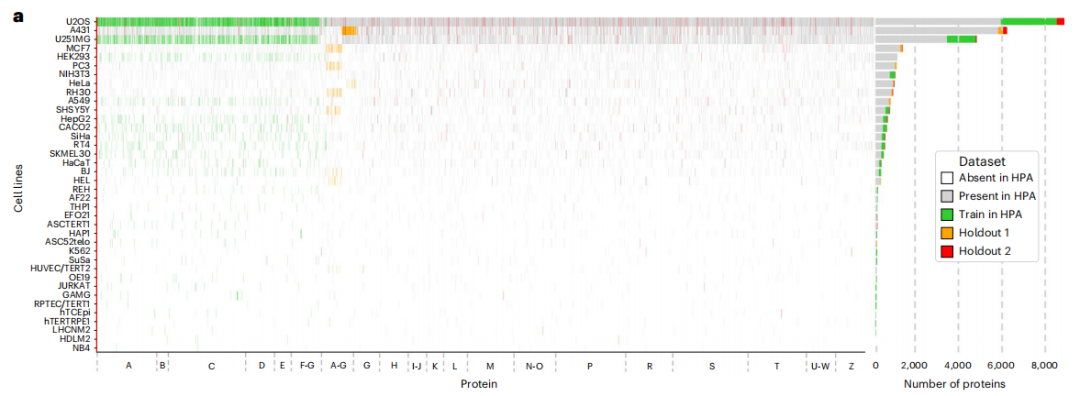
训练集(绿色),保留集 1 (橙色),保留集 2 (红色)
HPA 中未采用部分(灰色),HPA 中不包含部分(白色)
具体来说,训练数据集包含 340,553 个细胞数量,蛋白质变体共 8,086 种,对应 HPA 中 37 种细胞系中的 2,801 个基因,这些基因名称以字母 A-G 开头。另外,训练数据集中还额外包含了 10 个基因,包括 IHO1、IMPAD1、INKA1、ISPD、ITPRID1、KIAA1211L、KIAA1324、LRATD1、SCYL3、TSPAN6。
保留数据集分为两部分:一部分为保留数据集 1,包含 36,552 个细胞,蛋白质变体由 9,472 种构成,对应 3,312 个基因(含训练集中的 2,801 个),名称同样以 A-G 开头,但来自不同的细胞系,与训练集无重叠。同时,保留数据集 1 进一步被拆分为两个部分,用作评估集和测试集,分别包含 11,050 和 25,502 个细胞;保留数据集 2 含有 24,007 个细胞,对应 515 个基因,其名称以字母表所有字母开头,即涵盖 A-Z,蛋白质变体共 556 种,来自完全未在训练集和保留数据集 1 中出现的新基因家族,可用于模型泛化能力的测试。
另需说明的是,BJ 细胞系图像被同时保留在了训练集和保留数据集 1 中。
在实验之前,研究团队对 HPA 中的图像进行了预处理,简单来说包含以下 5 步:
* 第一步,对每张图像向下采样 4 次,最终分辨率降至 0.32 μm/像素,以便减少计算量并去除高频噪声;
* 第二步,结合高斯模糊(σ=5)和 Otsu 阈值法从复杂背景中分离出细胞核的大致区域;
* 第三步,使用 remove_small_holes 函数,移除面积小于 300 像素的孔洞,然后将图像二值化,并去除小于 100 像素的噪声区域;
* 第四步,计算每个细胞核的质心,并以质心为中心,裁剪出 128 x 128 像素的区域作为单个细胞的 ROI;
* 第五步,通过强度归一化和噪声过滤,实现标准化数据分布,减少通道间干扰。
模型架构:结合蛋白质序列和图像表征预测蛋白质亚细胞定位
PUPS 模型主要由两个部分组成,一个用于从蛋白质的氨基酸序列中学习序列表示;另一个用于从靶细胞的标志性染色中学习图像表示,然后结合蛋白质序列表示和图像表示来预测蛋白质在靶细胞中的亚细胞定位。前者使模型能够推广到未知蛋白质预测,后者使模型具备捕获单细胞水平的变异性,实现了细胞类型特异的定位预测。如下图所示:
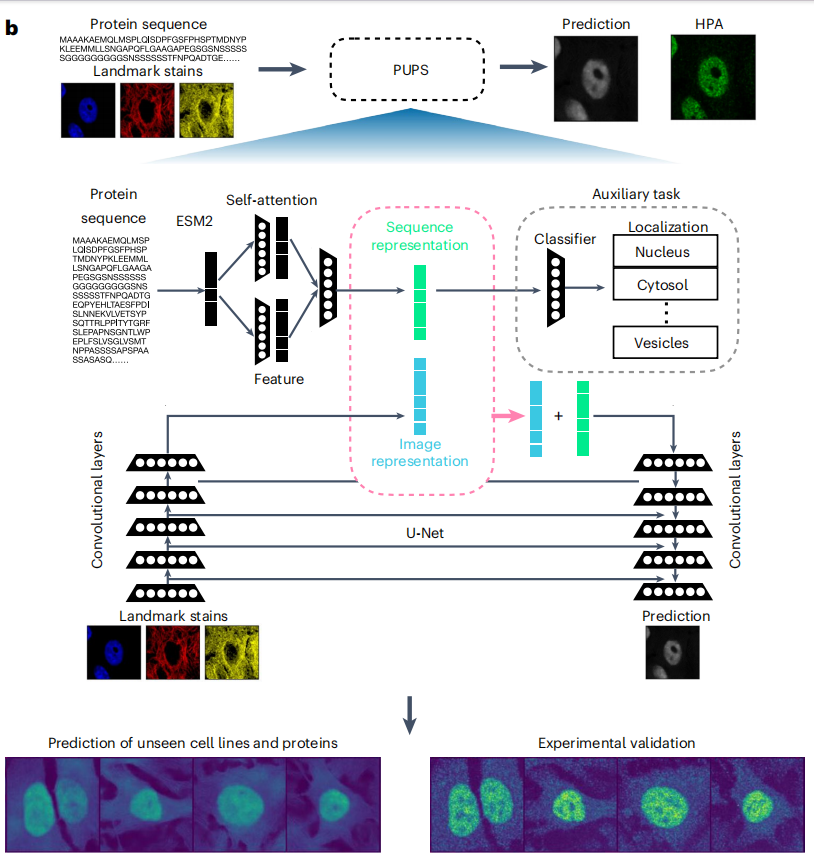
未知细胞系中未知蛋白质亚细胞定位演示
简单来说,PUPS 利用了预训练的 ESM-2(Evolutionary Scale Modeling)蛋白质语言模型提取蛋白序列特征,同时用卷积神经网络学习细胞的标志性染色图像特征,最终结合两部分信息预测蛋白质在靶细胞中的定位。需要说明的是,模型所有部分同时进行训练,有助于减少前置任务的分类损失,以及预测蛋白质图像与 HPA 中实验测量的蛋白质图像之间的差异。所有参数使用 Adam 优化器进行优化,学习率为 1e-4。
蛋白质语言模型
PUPS 通过使用语言模型、自注意力层以及一个辅助预训练任务来学习序列表征,然后根据学习到的序列表征对蛋白质定位进行分类。
具体来说,研究团队通过将 N 端 2,000 个氨基酸序列输入到预训练 ESM-2 模型中,获得特定蛋白质变体的初始表示,从而为每个氨基酸残基生成 1,280 维向量,残基少于 2,000 的变体采用零填充。这种序列长度截断是为了避免对序列长度高达数万个残基的少数蛋白质进行偏倚预测。如下图所示:
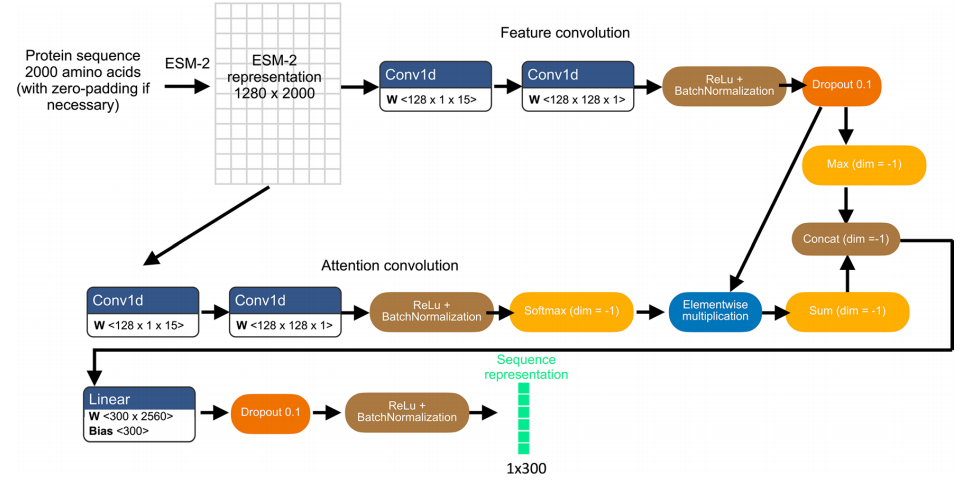
基于预训练 ESM-2 模型与轻量注意力层的蛋白质序列表征学习模型架构
为了使 ESM-2 表征适应于蛋白质定位预测,团队在后续采用了可分离卷积(separable convolutions)的轻注意力层,应用于 ESM-2 表示最终获得 300 维序列表征。这种蛋白质序列表示既用于预测定位标签的辅助前置任务,同时也用于与图像表示相结合的蛋白质图像预测。前置任务将蛋白质序列表示输入到一个全连接的神经网络层,以输入一个 29 维向量,表示 29 个亚细胞区室定位标签中的概率分布,然后利用 S 型激活(sigmoid activation)的二元交叉熵损失将前置任务输出结果与 HPA 注释的蛋白区室进行比较。
图像绘制模型
每个细胞的图像输入包含了细胞核、微管和内质网染色这 3 个标志性染色图像通道,其维度为 3 x 128 x 128,并以细胞核质心为中心。
图像编码通过 5 个可分离卷积层实现,最终维度 16 x 16 x 512。每个卷积层之后依次连接 leakyRelu 激活,批归一化以及 2D 最大池化层。蛋白质序列表示被拼接至细胞图像表示的所有空间维度,随后输入 U-Net 图像解码器,为每个输入通道学习不同权重。此外,模型中的空间维度加权机制允许图像表征的每个空间维度以不同权重与序列表征相结合。
解码器由 5 个可分离卷积层构成,生成 1 x 128 x 128 的图像输出,即对应细胞的蛋白质图像预测。然后将类似于图像分割 U-Net 的跳跃连接(skip connentions)添加在标志染色生成图像表示的编码层与同深度生成蛋白质图像预测的解码层之间。研究采用了均方误差损失函数训练模型,以最小化预测蛋白质图像与实验测量蛋白质图像之间的差异。
实验结果:实现单细胞级蛋白质亚细胞精准定位
为了验证模型的可行性和有效性,研究团队提出多项实验进行验证,PUPS 在多项任务中均表现出较好的性能,凸显了其多模型融合的优势。
预测细胞系间蛋白质定位的变异性
为了评估 PUPS 在定量分析蛋白质于细胞系间定位变异性方面的性能,研究团队通过计算蛋白质核内比例量化定位变异性,发现预测值与真实数据高度相关,Holdout 1 的 pearson 相关系数为 0.794,Holdout 2 的 pearson 相关系数为 0.878。如下图所示:
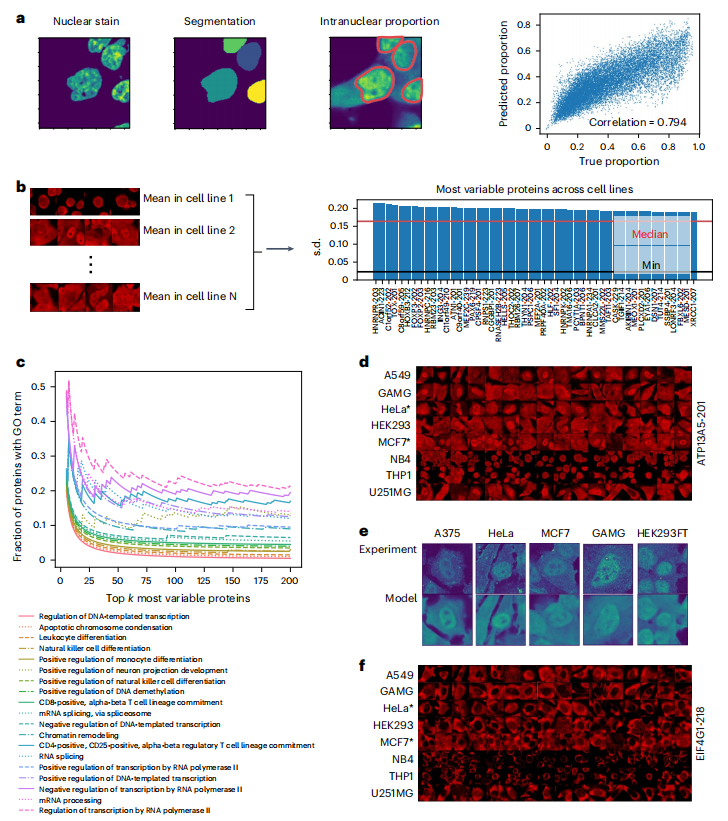
PUPS 精准预测不同细胞系间蛋白质定位的差异
随后进一步分析显示,细胞系间定位变化最大的蛋白质与转录、细胞分化和染色质调节等生物过程相关,如 ATP13A5 的实验验证证实了模型预测的准确性。此外,模型通过标志性染色捕捉细胞形态差异,无需细胞系标签即可推断蛋白质定位的细胞系特异性,为研究蛋白质功能的细胞特异性调控提供了新方法。
预测单细胞间蛋白质定位的差异性
为了评估 PUPS 对同一细胞系内单细胞间蛋白质定位变异性的预测能力,研究团队计算了每个细胞系中所有单细胞中蛋白质的核内比例方差,结果发现每种蛋白与细胞系对的单细胞变异性预测排名与真实数据高度一致,如 Holdout 2 中前 500 个高变异对重叠率超过了 60%,并且预测的核内比例分布与实际结果一致,排除了预测误差影响。
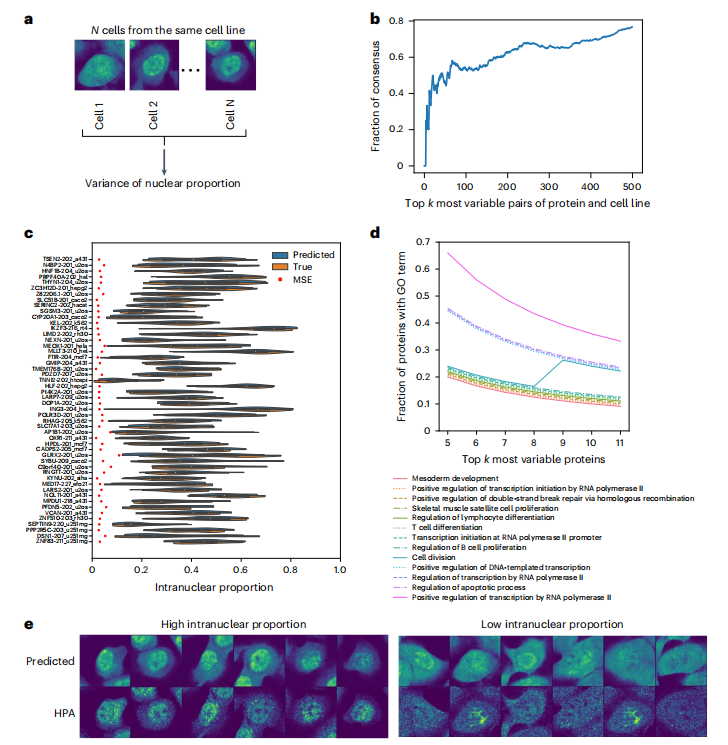
PUPS 可预测细胞系内单细胞中蛋白质定位的可变性
另外 Gene ontology(GO)分析表明,高度可变的蛋白质与细胞分裂、转录、双链断裂修复以及凋亡等过程有关。此外,模型通过细胞标志性染色图像捕捉形态等特征,表明了单细胞变异性不仅具有随机性,还与细胞形态特征相关,为解释单细胞异质性机制提供了新视角。
PUPS 在训练数据之外的新实验中的验证
为了验证 PUPS 在新的实验环境下预测蛋白质定位的泛化能力,研究团队选择了 9 种蛋白质在 5 个细胞系中进行验证。如下图所示:
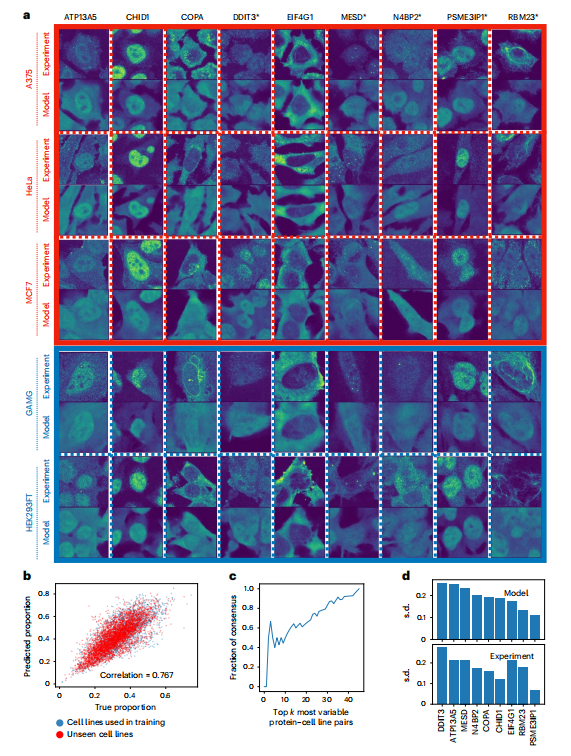
PUPS 在 HPA 之外的实验中预测蛋白质亚细胞定位的能力
ATP13A5、CHID1、COPA、MESD 和 RBM23 为细胞系间变异最大的蛋白,它们都有不同的 GO term;DDIT3 和 N4BP2 是细胞系内单个细胞中变异最大的蛋白;EIF4G1 和 PSME3IP1 是细胞系间变异最小的蛋白,前者预计主要位于细胞核外,后者预计主要位于细胞核内。5 个细胞系中,除 A375 外,其他 HeLa、MCF7、GAMG 和 HEK293FT 均包含在 HPA 中。
结果显示,PUPS 预测的蛋白质图像在视觉上与实验测量的图像相似。利用预测蛋白图像计算的每个单细胞的核蛋白比例与实验测量图像计算的比例密切相关,pearson 相关系数为 0.767。这表明,PUPS 可以用于定量预测以前没有实验测量或在训练图谱中使用的蛋白质的定位。
PUPS 学习到有意义的蛋白质和细胞表征
实验证明,PUPS 在未知蛋白质和细胞系中预测蛋白质定位的能力来自于学习到了蛋白质序列和细胞标志性图像的有意义表示。
研究团队绘制了对应于 12,614 个基因的 40,622 个蛋白质形态的蛋白质序列表示,具有相似定位的蛋白质往往具有相似的序列表示。为进一步证明模型能识别有意义的蛋白质序列模式以及预测定位,研究团队使用 Positional Shapley 方法计算了特定蛋白质中每个氨基酸残基对预测各细胞区室标签预测的重要性,如成功解释了 N4BP2 核定位的预测变异性,也与 CUE 结构域通泛素结合可能改变亚细胞定位的报道相符。
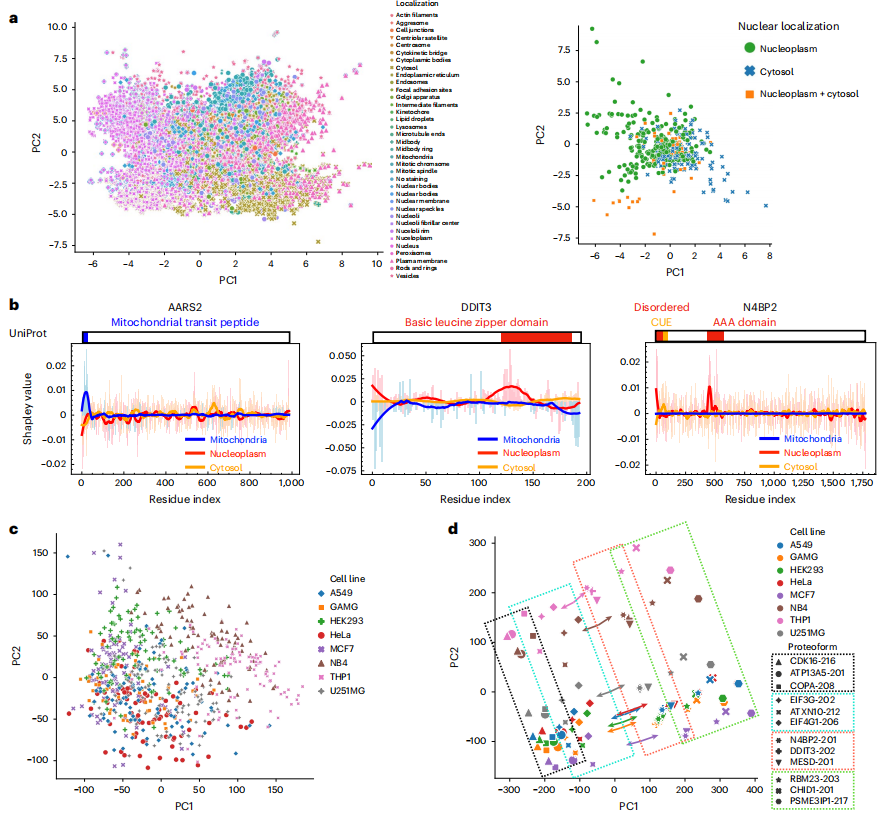
PUPS学习有意义的蛋白质和细胞表征
除此了识别有意义的蛋白质序列基序外,研究团队进一步表明了 PUPS 从细胞标志性染色中学习单细胞的有意义表征。其将从标志性染色中学习到的单细胞图像表示可视化,发现即使细胞系标签没有输入到模型当中,同一细胞系的单细胞也具有相似的图像表示。蛋白质和细胞标志性图像的联合表示保留了细胞系和蛋白质之间的分离,而每个细胞系内的不同蛋白质在不同细胞系之间的顺序相似。给定联合表示空间中每个细胞系的质心,从质心到特定蛋白质的向量在所有细胞系中大部分是平行的,即在给定序列表示的情况下,预测特定蛋白质的图像需要再表示空间中以相同方向移动,而不管细胞系是什么,这解释了 PUPS 通过学习有意义的蛋白质和细胞图像表示来推广到未知蛋白质和细胞系的能力。
此外,PUPS 还能预测致病突变对蛋白质定位的影响。例如,针对核编码的线粒体蛋白 SDHD 和 ETHE1 的突变研究表明,SDHD 突变会导致其核定位比例增加,这与疾病中核基因组不稳定的机制一致;ETHE1 突变则显示胞质定位比例升高,与已知的核 – 胞质穿梭异常相关。这些结果表明,PUPS 可通过分析序列变异对定位的影响,为疾病机制研究提供新线索。
蛋白质亚细胞定位预测新解
正如上述所言,蛋白质亚细胞定位预测在生物信息学和生物学研究中都具有重大意义,PUPS 提供了一种融合多模态信息的思路,为该领域的研究画上了浓墨重彩的一笔。与此同时,该领域的研究经过数十年的发展,其成果也早已是百花齐放。
爱尔兰都柏林大学的团队在 Computational and Structural Biotechology Journal 杂志上发表了一项研究,其中围绕蛋白质亚细胞定位预测介绍了多种计算方法,包括基于序列、注释、混合及元预测等类别,同时文章还按真核生物、原核生物、病毒及多类别对亚细胞定位预测工具进行了分类介绍,真核生物预测工具如 mLASSO-Hum、DeepPSL 等,原核生物预测工具如 PRED-LIPO 等。通过设计涵盖 7 个主要领域及 28 个子分类的机器学习和深度学习分类图,该研究提供了单类别和多类别预测工具分类法,从而方便用户查找方法、预测工具。论文以「Protein subcellular localization prediction tools」发表。
* 论文地址:
https://www.sciencedirect.com/science/article/pii/S2001037024001156
复旦大学生物医学研究院杨力研究组与上海人工智能实验室董楠卿研究组合作,于 4 月 12 日在 Briefings in Bioinformatics 杂志在线发表了题为「Deep Generative Model for Protein Subcellular Localization」的研究论文。研究同样基于 ESM2 蛋白质大语言模型及 U-Net 框架,开发了具备多模态处理能力的生成式深度学习模型 deepGPS。
据介绍,deepGPS 能够接收蛋白质序列及细胞核图像作为输入,并生成蛋白质定位的文本标签及分布图像,是一种支持蛋白质亚细胞定位预测的新型「文生图」(text-to-image)多模态模型。
* 论文地址:
https://doi.org/10.1093/bib/bbaf152
随着人工智能与生物学研究的融合加速,相关的创新性实验也在不断涌现,并逐渐打破传统方法的弊端,实现「两全其美」甚至「十全十美」的表现,从而推动生物信息学的快速发展。



戳“阅读原文”,免费获取海量数据集资源!
(文:HyperAI超神经)