

作者:郑伟
编辑:十九,李宝珠
本文获得郑伟教授授权发布,转载请联系本公众号获得授权,并标明来源
南开大学郑伟教授以「AlphaFold3 王座未稳,来自学术界的反超:基于深度学习的生物大分子及其互作的三维结构预测」为主题,分享了 AlphaFold 的局限和未来优化方向、以及学术界还有哪些值得探讨的算法和研究课题等。
近年来,在深度学习等 AI 技术的辅助下,蛋白质结构预测领域发展迅猛,2024 年 10 月份,因为 AlphaFold,DeepMind 的 Demis Hassabis、John M. Jumper 获得了 2024 年诺贝尔化学奖,然而,这并非代表 AlphaFold 已经无可替代,其他优秀算法仍然值得挖掘。
在「Meet AI4S」系列直播的第六期,HyperAI超神经有幸邀请到了南开大学统计与数据科学学院教授郑伟教授,他以「AlphaFold3 王座未稳,来自学术界的反超:基于深度学习的生物大分子及其互作的三维结构预测」为主题,向大家分享了 AlphaFold 的局限和未来优化方向、以及学术界还有哪些值得探讨的算法和研究课题等。
* 关注公众号,回复「Meet AI4S 第六期」获取演讲 PPT
HyperAI超神经在不违原意的前提下,对其深度分享进行了整理汇总,以下为演讲实录。
点击查看完整演讲回放 ⬇️
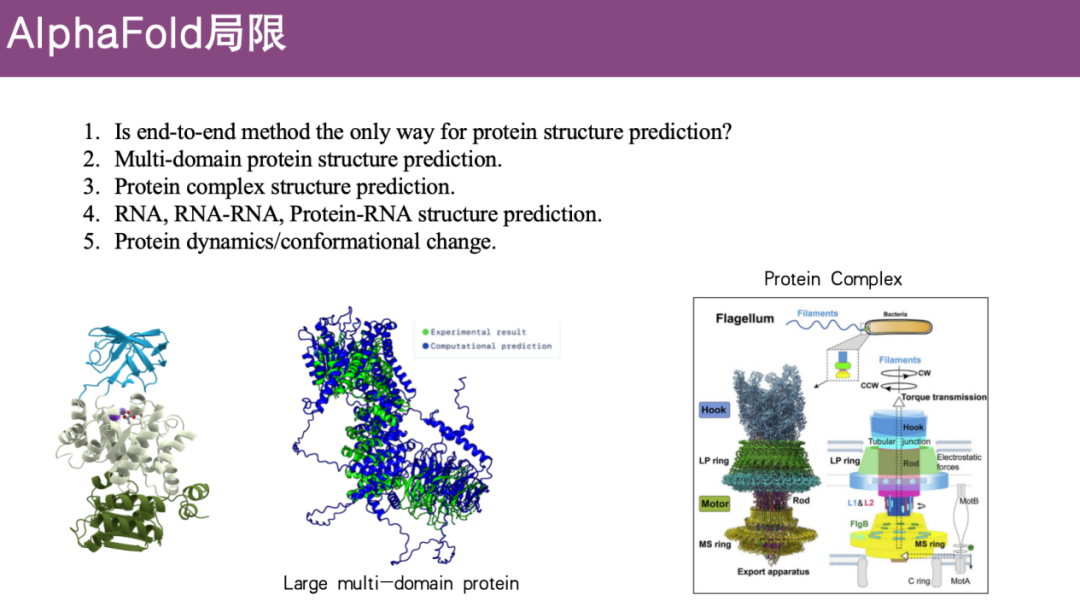
AlphaFold 的局限
蛋白质是生命活动的基石,预测蛋白质三维结构对于了解生物学功能至关重要。虽然 DeepMind 推出的 AlphaFold 2 将蛋白质结构预测提升到了一个新高度,但这并不意味着,AlphaFold 2 的端到端框架已经解决了所有蛋白质结构预测问题。
首先,以 AlphaFold 2 自身为例,它还存在很多局限:
* 精度有待提升
官方报道 AlphaFold 2 能够以 90% 以上的精度预测结构,但实际 task 并不能达到这么高。
* 多域蛋白质结构预测受限
AlphaFold 2 在单结构域蛋白上预测较好,但复杂的多结构域蛋白,结构域之间比较 flexible,预测精度并不好。
* 蛋白质复合物结构预测受限
蛋白质通常需要与其他蛋白质形成复合物发挥作用,但 AlphaFold 2 的最初版本并未涉及该问题。
* RNA 结构预测、RNA-RNA、蛋白质-RNA 结构预测受限
和上述一样,其初始版本未曾涉及这些问题。
* 蛋白质动态/构象变化预测受限
实验解析手段通常只能捕捉到某一时刻的结构状态,但蛋白质在其生物体内并非静态存在,不同时间点的结构可能各不相同,而这些问题 AlphaFold 2 尚未解决。
进一步地,虽然 DeepMind 已经迭代了 AlphaFold 3,我们也都知道其在蛋白质单体结构预测方面表现出色,但它在复合物、核酸和小分子预测上的精度仍有待提升。因此,下一代 AlphaFold 或许会增加其他功能的预测模块,比如考虑到现有模型主要用于处理静态结构,将会探索分子动态过程,预测蛋白质的变构。此外,还可能涉及蛋白质设计领域,将整个预测流程逆转。
因此,即便有了 AlphaFold,整个学术界仍有大量工作要做。
除了 AlphaFold,还有什么值得挖掘的方法?
过去,我们解析蛋白质三维结构的方式主要有 x 射线、核磁共振 (NMR)、冷冻电镜,由于实验解析蛋白质结构的难度和高昂成本,一些团队可能需要花费数月至数年的时间才能解析一个蛋白质三维结构。于是,人们开始探索更经济和快速的方法,也就是通过算法预测蛋白质结构。
我们知道,蛋白质主要由 20 种氨基酸组成,通常用英文字母来表示,氨基酸分子里也含有很多原子。因此蛋白质结构预测问题可以概括为:输入一个由这些字母组成的氨基酸字符串,使用计算算法去预测蛋白质序列中的每个氨基酸中的每个原子的三维空间坐标 (x, y, z) 。
纵观整个蛋白质结构预测的发展历程,不同阶段出现了多种代表性算法,如比较建模或同源建模、分子动力学模拟(MD)、Threading 算法、从头预测 (de novo prediction)、基于深度学习预测接触图的结构预测算法等。主要介绍如下:
* 比较建模或同源建模
该方法基于生物进化原则,认为如果序列相似性高,那蛋白质的结构和功能也较为相似。因此,可以首先获取未知蛋白质的氨基酸序列,然后在 PDB 数据库中通过序列比对找到序列相似度高的、已经解析的蛋白质结构模板,通过迁移或比对来预测未知蛋白质的结构。
*PDB 数据库收录了该领域已经解析的蛋白质的结构

* 分子动力学模拟
基本思路是根据蛋白质的氨基酸序列随机生成一个初始三维结构,赋予每个原子随机坐标,通过调整原子位置,再根据预先构建好的物理能量力场,计算不同时刻蛋白质的状态能量,能量最低的结构就是合理的蛋白质构象。

* Threading 算法
与同源建模相似,其不同点在于,虽然序列相似性高的蛋白质结构往往相似,但结构相似的蛋白质其序列相似性可能很低,这类蛋白无法在 PDB 数据库中找到合适的模板信息。于是研究人员提出了 profile 概念,在收录的同源序列基础上,利用多重序列对比 (MSA),以对齐两个蛋白质 profile 的方式来对齐不同的氨基酸。
也就是说,即使两个氨基酸序列不同,但它们的 profile 相似,即可认为两者的结构类似,以此寻找模板。

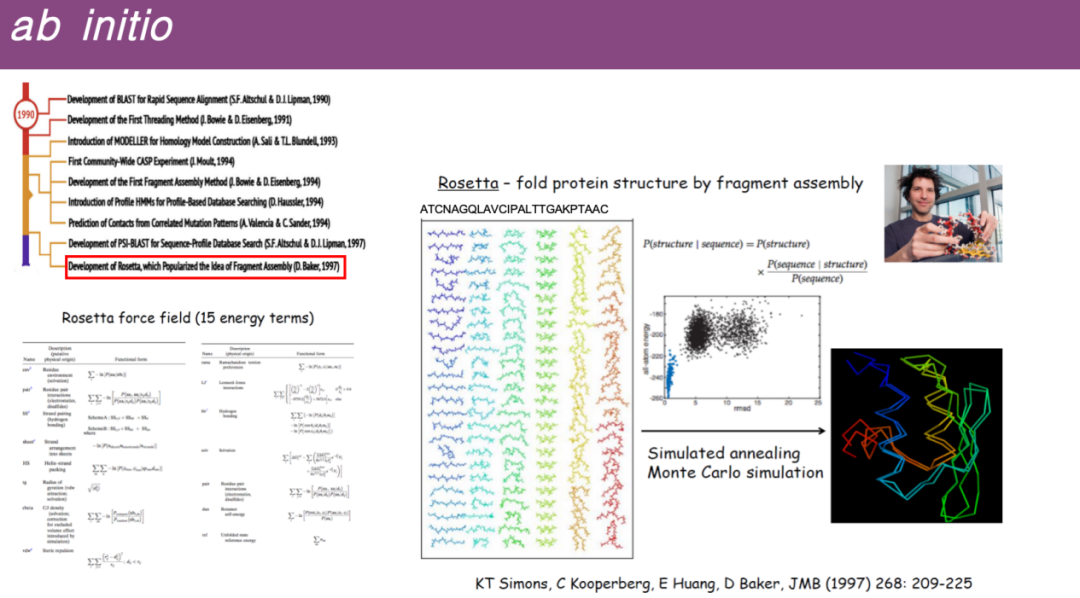
* 从头预测 (de novo prediction)
一些蛋白质在数据库里并不一定能找到相似的结构,于是研究人员试图通过将整个蛋白质序列分解成较短片段,在数据库中寻找这些小片段的模板,再将这些小片段模板组装成完整的三维结构,以此进行预测。
具体来讲,华盛顿大学的 David Baker 教授开发了 Rosetta 软件,其主要原理是将蛋白质序列分解为许多小片段,随机组装这些片段后,再利用分子动力学模拟中开发的能量函数进行优化,通过类似于动态模拟和能量最小化原则来进行结构预测。
* 接触图
主要思想是将蛋白质的三维结构转化为二维图。利用蛋白质三维结构信息,即用所有空间点的坐标位置来计算不同氨基酸之间的距离,假定两个氨基酸距离小于一定的阈值时形成 contact,反之,则不形成 contact,用此定义将三维结构压缩为二维图。进一步地,这种二维接触图的信息可以重构蛋白质的三维结构。
具体来讲,研究人员开发了许多基于深度学习的方法,其核心思想是,首先构建多重序列比对 (MSA),观察氨基酸 i 和 j 里的 profile 的协同进化信息,因为这种协同进化的氨基酸在空间上往往距离很近,会形成接触。随后,将共进化信息作为特征输入到深度学习网络进行训练,进而预测蛋白质的接触图,恢复蛋白质三维结构。
例如,郑伟教授团队之前开发了一种名为 C-I-TASSER 的算法,就是目前基于接触图预测蛋白质结构的一种常用方法。

最后,AlphaFold 集成了上述很多算法的基础原理,成功构建了端到端的框架,可以直接输入蛋白质序列,随之输出结构。
以团队成果为例,探讨学术界的反超机会
蛋白质结构预测对生物医药领域有巨大影响,比如,目前郑伟教授团队开发的算法就涉及到预测未知病毒蛋白结构(新冠病毒)、辅助冷冻电镜解析蛋白质结构、帮助生物学家理解蛋白质进化功能、抗体筛选等。
此外,如下图所示,团队开发的所有蛋白质单体和复合物结构预测算法均已转化为自动服务器算法,并在课题组网站上公布,其算法已经服务于全球 100 多个国家的 9 万多名用户,欢迎大家使用。
*总项目地址:
https://seq2fun.dcmb.med.umich.edu/DMFold/

蛋白质单体结构预测方法 D-I-TASSER
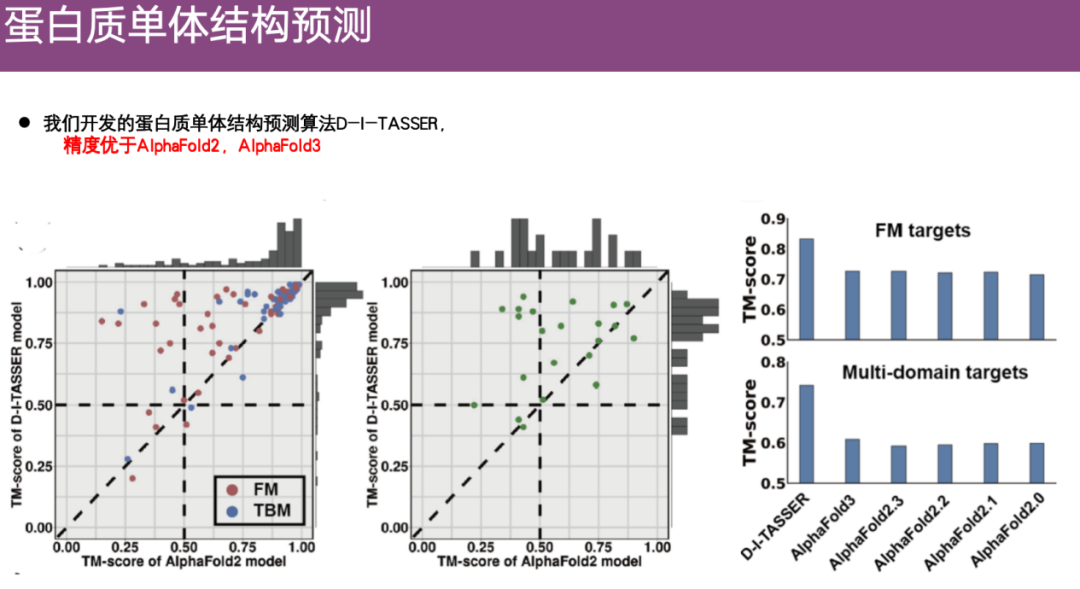
蛋白质单体结构预测问题一直备受关注,在 AlphaFold 2 之前,郑伟教授团队就已经在做基于接触图的结构预测研究。AlphaFold 2 出现后,团队开始思考,能否将 AlphaFold 2 预测的接触图等空间约束整合到之前开发的算法中。于是基于空间约束、宏基因组、统计能量函数等,团队开发了蛋白质单体结构预测算法 D-I-TASSER,经过优化,该算法展现了良好的效果。
如下图右侧案例所示,红色表示 D-I-TASSER 预测的蛋白质结构,蓝色表示实验解析的结构。可以看到,D-I-TASSER 预测的结构与实验解析的结构非常相似,相比之下,AlphaFold 2 预测的结构与实验结构即使对齐后也存在明显差异,其预测精度略低。

此外,在多个蛋白质数据集进行评测。如下图右侧所示,预测单结构域和多结构域时,D-I-TASSER 的预测精度高于 AlphaFold 2,甚至高于 AlphaFold 3。
为了确保评估的权威性,团队不仅进行内部评测,还参加了领域内权威的比赛——CASP。
CASP 比赛被誉为该领域的奥林匹克竞赛,主要是为了标准化蛋白质结构预测的评估方法。因为蛋白质三维结构预测算法种类繁多,各实验室也开发了各自的算法,由于评测的数据集和方法可能不同,每个课题组通常都声称自己的方法是世界上精度最高的。为了解决这一混乱局面,CASP 比赛应运而生。
截至去年,CASP 已成功举办 16 届,持续了 32 年,吸引了众多权威团队参与,比如 David Baker 教授团队、DeepMind 团队等。
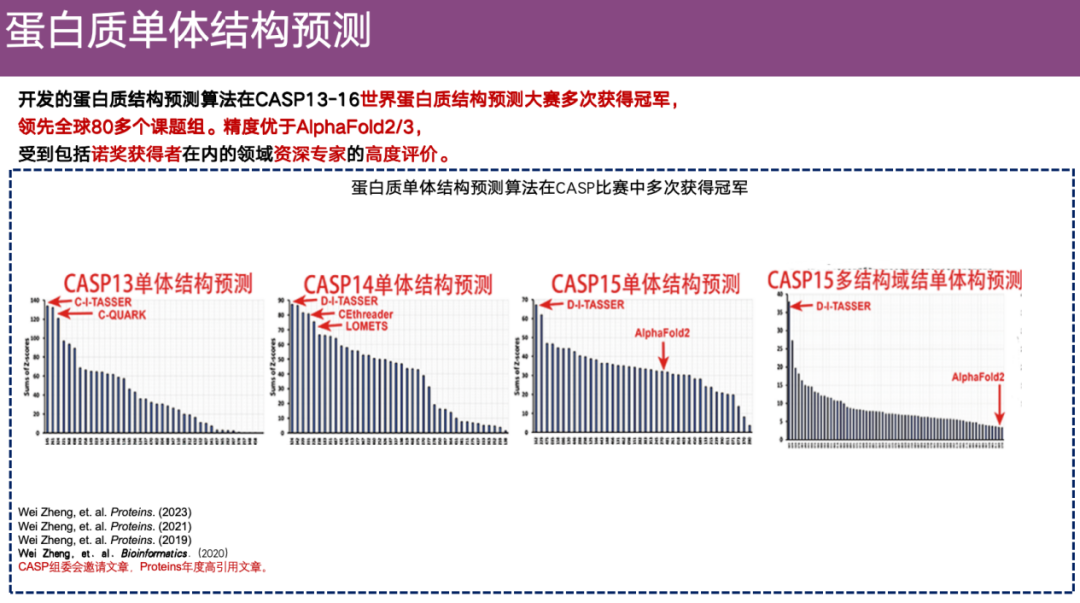
D-I-TASSER 及其前身算法多次参与 CASP 比赛,CASP 13-CASP 15 期间,该方法在蛋白质单体结构预测领域一直处于领先位置,在 CASP 15 中,D-I-TASSER 算法还参加了多结构域评测,整体精度优于所有参赛课题组。
蛋白质复合物结构预测方法 DMFold
复合物结构预测的主要挑战在于预测两个蛋白质之间的相对扭转,这可以通过共进化信息去分析。
比如,通过对两个蛋白质的单体进行多重序列比对 (MSA) 构建,并基于一些连接手段将两个 MSA 合并成一个 MSA,并利用这两个 MSA 之间氨基酸的共进化关系来推断不同蛋白质间氨基酸的距离,还可以把共进化信息整合到深度学习框架中,预测两个蛋白质间的相对扭转。
对此,郑伟教授课题组开发了 DeepMSA 和 MetaSource 算法,用于构建更深层次的多重序列比对。此外,团队还利用深度学习、宏基因组等开发了蛋白质复合物结构预测算法 DMFold。

如上图最右侧案例所示,上方是通过实验解析获得的真实结构,下方左侧是 DMFold 预测的结构,右侧是 AlphaFold 2 预测的结果。可以看到,AlphaFold 2 预测的结构较为混乱,并且出现了异常的触手状延伸。相比之下, DMFold 的预测结构与实验结构高度相似,说明在复合物结构预测方面, DMFold 算法优于 AlphaFold 2。
此外, DMFold 还在大体系蛋白质—蛋白质复合物、纳米抗体-抗原复合物、点突变引起的构象变化等方面,表现出较高的精度。在 CASP 15 比赛中, DMFold 的整体排名远高于 AlphaFold 2,在 CASP 16 中, DMFold 也优于 AlphaFold 3。

RNA-RNA 复合物结构预测方法 ExFold
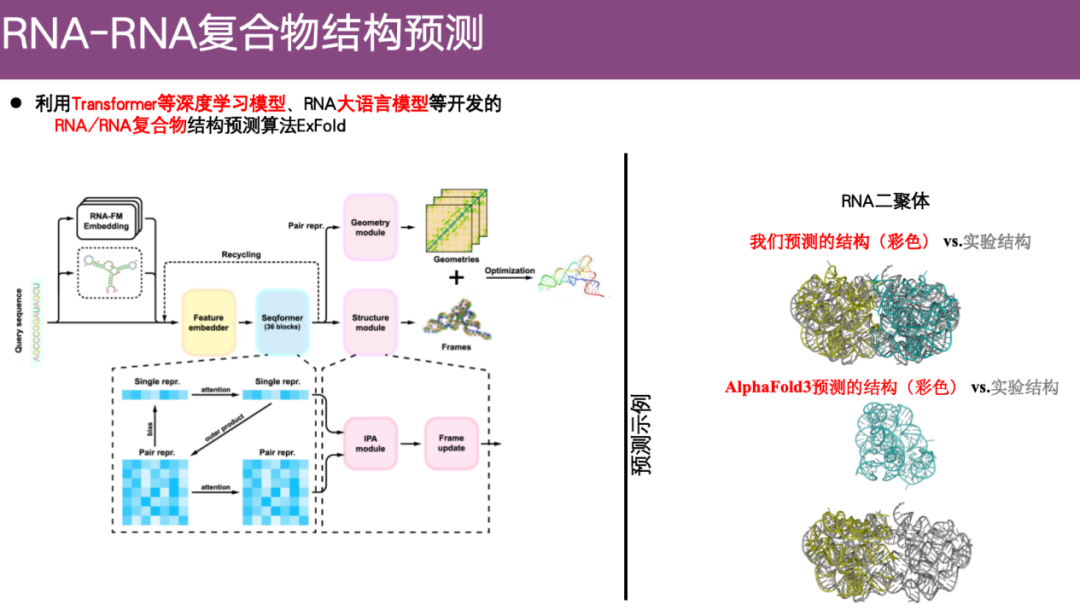
团队近年来开始关注 RNA 结构预测问题,比如,利用 Transformer 等深度学习模型、RNA 大语言模型等开发了 RNA/RNA 复合物结构预测算法 ExFold。
如下图右侧案例所示,灰色部分是实验结构,彩色部分是预测的结构。可以看到,用 ExFold 方法,这两个结构对齐效果良好,相比之下,AlphaFold 3 预测的两个 RNA 分子之间甚至未形成接触,几乎可以认为是完全错误的。
团队还通过一个较大的数据集与 AlphaFold 3 进行了精度比较,如下图左侧所示,Y 轴表示 ExFold 的预测精度,X 轴表示 AlphaFold 3 的预测精度。可以看出,ExFold 的优势还是较为明显的。
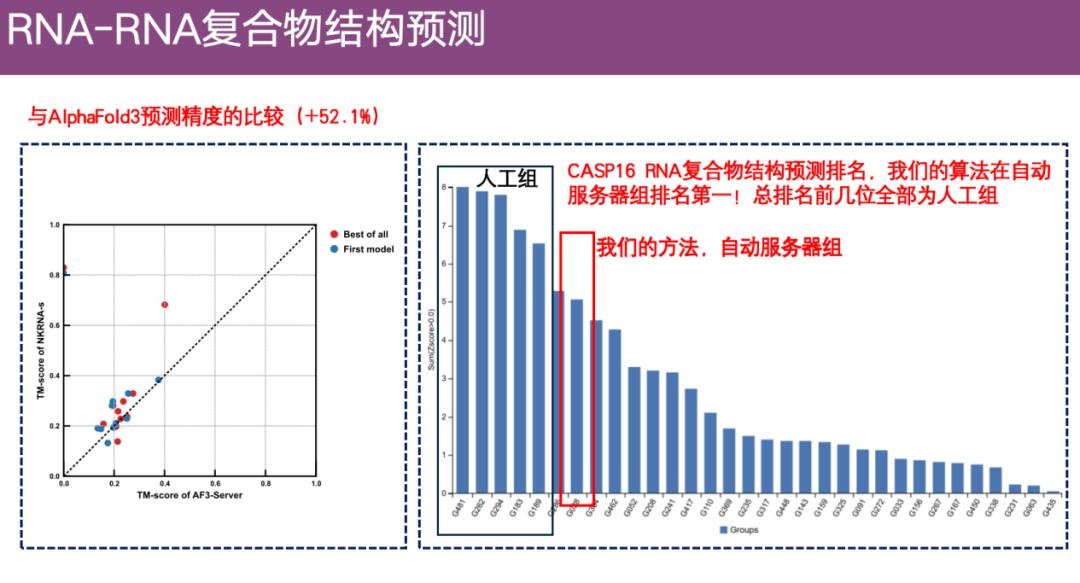
另外,在 CASP 16 的 RNA 复合物结构预测比赛中,ExFold 虽然总体排名不是第一,但在所有自动算法(服务器算法)中,其排名最高。
* CASP 比赛分为自动组和人工组。自动组要求在 3 天内全自动提交预测结果,不允许任何人工干预;人工组有 3 周时间,允许加入专家经验和人工调整。
蛋白质-RNA 复合物结构预测方法 DeepProtNA
关于蛋白质-RNA 复合物结构预测问题,团队利用 Transformer 等深度学习模型及最近流行的大语言模型,开发了一种新的结构预测算法——DeepProtNA。
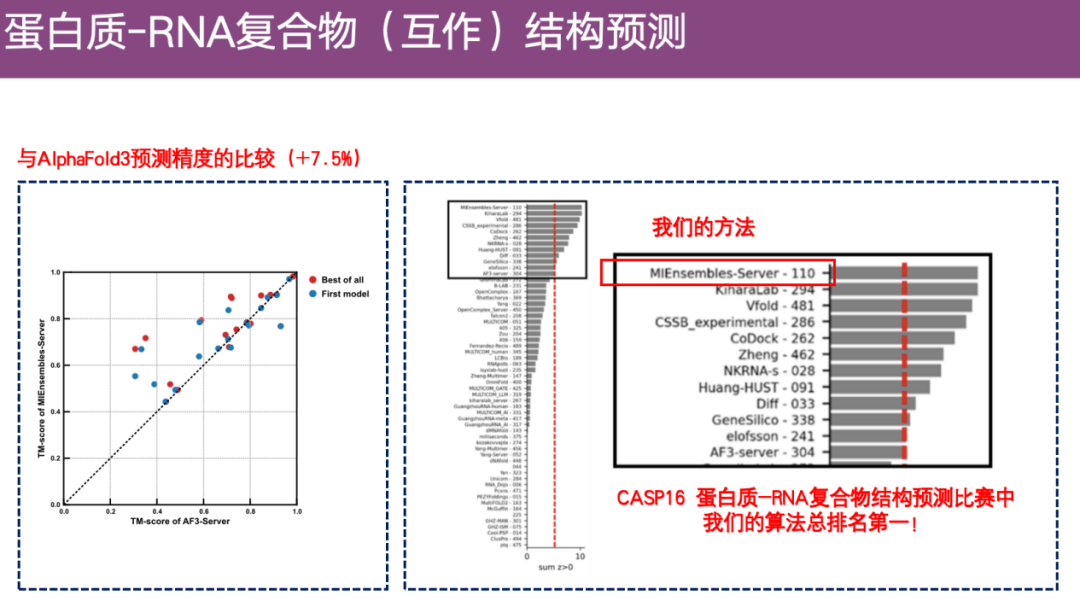
如下右侧案例所示,在抗体-RNA 复合物中,彩色代表 DeepProtNA 的预测结果,而灰色为实验结构。对齐后可以发现,DeepProtNA 的预测结构与实验结构高度重合(灰色与彩色重合),特别是在抗体蛋白和抗原 RNA 的接触面上,预测精度非常高。相比之下,AlphaFold 3 的预测结构与实验结构几乎无法重合,预测效果较差。

此外,DeepProtNA 比 AlphaFold 3 的预测精度大约高出 7.5 个百分点,在 CASP 16 的服务器组比赛中,排名第一。
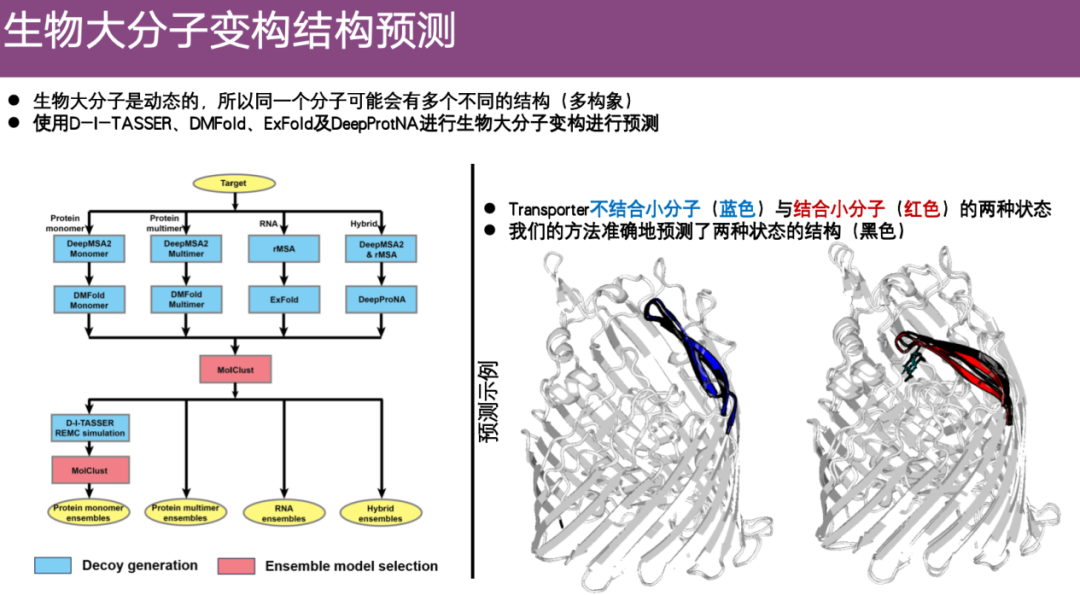
生物大分子变构结构预测方法 EnsembleFold
团队还关注生物大分子变构结构预测问题。大分子多构象问题的输入是一个蛋白质序列,输出是该蛋白质在不同状态下的多个关键帧,这意味着相对于静态预测算法,需要从单个氨基酸序列预测出多个不同的结构,这些结构代表整个动态过程中的关键帧。这是一个当前领域内关注度高但预测难度较大的课题。
通过整合之前开发的方法并对大分子变构问题进行优化,课题组开发了一些聚类算法,并最终形成了一个名为 EnsembleFold 的算法。
如下图右侧案例所示,展示了蛋白质结合小分子后的构象变化,蓝色表示未结合小分子时的实验结构,红色表示结合绿色小分子后发生倾斜和变构,团队基于输入的蛋白质序列预测出了两个结构,即黑色部分。可以看到,不结合小分子时 EnsembleFold 的预测结构与真实结构非常吻合,结合小分子后,EnsembleFold 也能很好地 fitting 到实验结构上。因此,EnsembleFold 在生物大分子变构预测方面表现出极高的准确性。
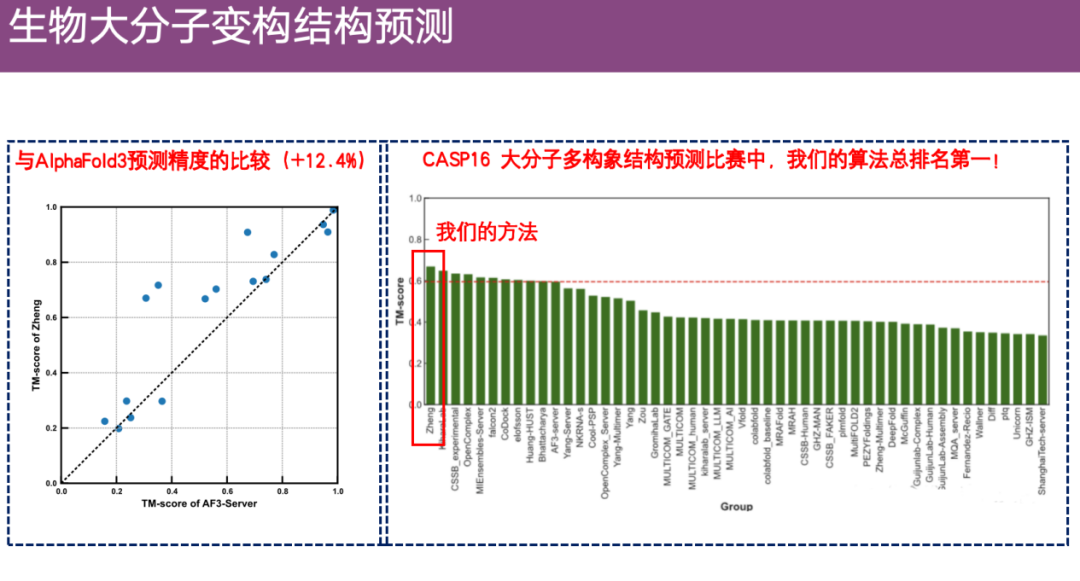
与此同时,与 AlphaFold 3 对比后发现,EnsembleFold 的精度高出约 12.4%。在 CASP 16 的所有大分子构象比赛中,其排名第一。
一个有趣的例子是,团队在 CASP 中对噬菌体 DNA 整合酶变构过程进行预测。如下图所示,噬菌体的氨基酸序列用 P -P’表示,细菌的遗传物质序列用 B-B’ 表示,通过动态过程,噬菌体 DNA 整合酶将噬菌体的遗传物质 P’ 整合到细菌的遗传物质B中形成 B-P’,构象产生变化。
团队使用算法对这种多构象变化进行了预测。左侧展示了实验结构,上部是未整合状态(构象 1),下部是整合后的状态(构象 2)。可以看到,课题组的预测能够准确反映这两种不同的构象。
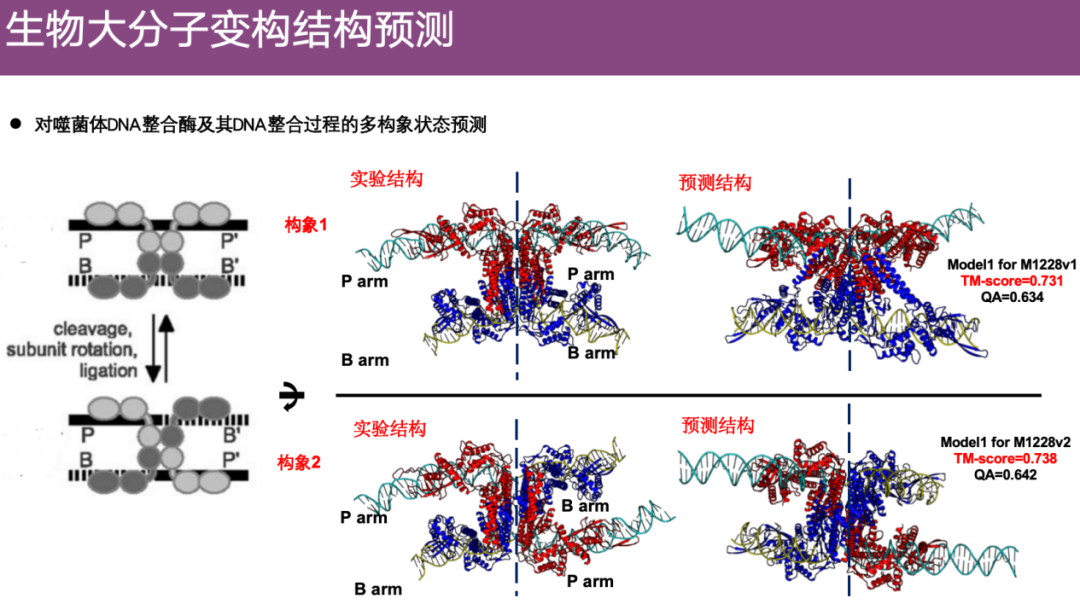
值得一提的是,在 CASP 16 比赛中,参赛者仅获得序列信息,并不清楚具体的生物学过程或构象变化细节,但郑伟教授团队通过预测成功还原了整个生物学过程,赛后总结时,评审委员也对此表示惊讶。
课题组招新
南开大学统计与数据科学学院郑伟教授长期致力于蛋白质等生物大分子结构、功能及互作的预测研究,并主导开发了多款精度优于 AlphaFold2/3 的蛋白质单体、蛋白质复合物、核酸及复合物、蛋白质核酸复合物结构预测算法,及结构评估算法等,在世界蛋白质结构预测大赛 (CASP) 多项比赛中多次获得冠军 (CASP13-16),领先全球 80 余个学术/工业界课题组。
其所在的南开大学统计与数据科学学院的生物信息学团队正在招募新成员,如果你对计算结构生物学或生物信息学、数据科学感兴趣,无论是硕士、博士还是博士后,都非常欢迎加入郑伟教授所在的团队。
感兴趣的同学可以通过以下方式联系郑伟教授:
* 邮件:jlspzw@nankai.edu.cn
* 微信:18622152765



戳“阅读原文”,免费获取海量数据集资源!
(文:HyperAI超神经)