
作者:哇塞
编辑:李宝珠
转载请联系本公众号获得授权,并标明来源
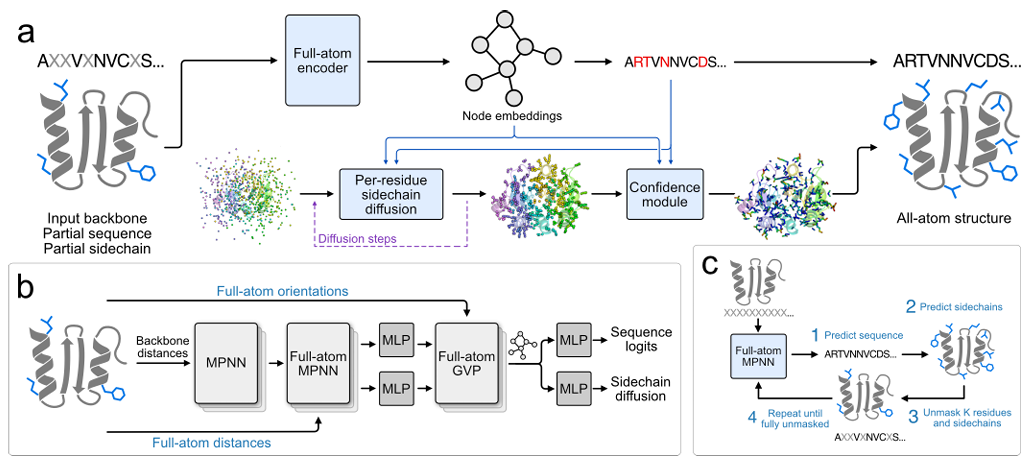
斯坦福大学的团队联合加州帕洛阿尔托市 Arc 研究院共同提出全新蛋白质序列设计方法 FAMPNN(Full-Atom MPNN),能够显式地建模每个氨基酸残基的序列身份和侧链结构。模型采用基于图神经网络的消息传递架构,结合改进的 MPNN 和 GVP 模块进行全原子编码,能够同时处理蛋白质的主链和侧链信息。
蛋白质侧链构象(Protein sidechain conformation),是指蛋白质中氨基酸残基的侧链在三维空间中的具体空间排布方式。研究蛋白质侧链构象可以帮助人们理解蛋白质结构和功能间的关系,在蛋白质工程、药物设计等领域都极具应用价值。然而,当前基于深度学习的蛋白质序列设计方法主要以固定主链蛋白质序列设计为主,其大多无法在序列生成过程中对蛋白质侧链构象进行建模,仅是基于主链几何结构和已知的氨基酸序列标签推断关键侧链的相互作用,而忽略了蛋白质侧链构象在蛋白质中的作用。
为了弥补上述空白,来自斯坦福大学的团队联合加州帕洛阿尔托市 Arc 研究院,共同提出了一种新的蛋白质序列设计方法 FAMPNN(Full-Atom MPNN),能够显式地建模每个氨基酸残基的序列身份和侧链结构。该模型采用基于图神经网络(GNN)的消息传递架构,结合改进的 MPNN(Message Passing Neural Networks)和 GVP(Geometric Vector Perceptron)模块进行全原子编码,能够同时处理蛋白质的主链和侧链信息。研究表明,FAMPNN 通过显式建模全原子结构,可以显著提高蛋白质序列设计的质量和实验预测的准确性。
研究成果以「Sidechain conditioning and modeling for full-atom protein sequence design with FAMPNN」为题,入选 ICML 2025。
研究亮点:
* 研究引入了一种结合交叉熵和扩散损失目标的方法,对残基的离散序列同一性和连续侧链结构的每个标记分布进行建模
* 研究实现了一种轻量级的迭代采样方法,用于从联合分布中生成样本,并利用改进的 MPNN 和 GVP 层进行全原子编码
* 研究证实,FAMPNN 通过显式建模全原子结构,能够有效改进序列设计和实验蛋白质适应性预测的准确性

论文地址:
https://go.hyper.ai/JUJDq
关注公众号,后台回复「FAMPNN」获取完整 PDF
更多 AI 前沿论文:
https://go.hyper.ai/owxf6
数据集:多样化数据集优化模型训练与评估
为了保证模型的有效性和可靠性,研究团队采用了复杂的多数据集进行训练和评估:
研究主要使用了 CATH 4.2 的 S40 数据集,该数据集是从蛋白质数据库(PDB)中提取的结构域的精选集,去除了同源性超过 40% 的冗余结构域,以 8:1:1 的比例划分为训练集、验证集和测试集。
PDB 数据集基于整个 PDB 数据库构建,包含截止 2021 年 9 月 30 日发布的结构,研究人员在蛋白质链水平上按照 40% 的序列同源性对蛋白质进行了聚类处理,优先保留多链蛋白质示例,用于训练模型以学习设计多链蛋白质。
CASP13、14、15 数据集主要用于评估模型在侧链 Packing 表现。研究团队使用 MMseqs2 建议搜索从训练和验证数据集中去除了所有与 CASP13、14、15 数据集中同源性超过 40% 的序列,然后通过预测侧链与真实侧链之间的平均均方根偏差(RMSD)来衡量侧链 Packing 性能。
SKEMPlv2 数据集用于评估模型对蛋白质-蛋白质结合亲和力的预测能力。该数据集整理了数百种蛋白质-蛋白质相互作用中数千个序列变体的实验测量结合亲和力,经过处理后,最终得到一个规模大小为 6,649 个数据点的数据集。
S669、Megascale、FireProtDB 数据集用于评估模型对蛋白质稳定性的零样本预测能力。这些数据集包含了对多种天然蛋白质稳定性变化的实验测量值(△△G),是稳定性预测器广泛使用的基准数据集。其中 Megascale 数据集为其精选、去重版本,研究团队将训练集、验证集和测试集合并为单个数据集,最终得到一个包含 272,712 个实验数据点的数据集,涉及 298 种不同的蛋白质。FireProtDB 数据集包含 100 种独特蛋白质的 3,438 个单突变的自由能变化,在经过处理后最终使用了其中 3,420 个示例。S669 数据集包含 94 种蛋白质的 669 个单突变的实验测量值,由于存在非标准氨基酸,该数据集中剔除了 4 个变体。
CR9114、CR6261、G6 数据集用于评估模型在抗体-抗原结合亲和力预测方面的性能。其中,CR9114 数据集中包含了 16 种氨基酸替换的所有可能组合。CR6261 数据集中包含了 11 种氨基酸替换的所有可能组合,分别共有 65,536 条和 2,048 条序列。G6 数据集共有 4,275 个与 VEGF-A 结合的数据点。
同时看懂蛋白质序列和侧链结构的智能工具
本次研究的核心目标是让模型同时学习蛋白质序列和侧链构象,为此,研究团队在序列一致性基础上采用掩模语言建模(Masked Language Modeling)对 FAMPNN 进行训练。训练采用端到端的方式,结合分类交叉熵损失(用于序列预测)和扩散损失(用于侧链构象预测),使得模型能够基于部分已知序列和侧链坐标,同时恢复被掩模的序列及其对应的侧链构象。
FAMPNN 训练策略
采样方面引入了类似 MaskGIT 的迭代采样策略,从序列和侧链完全被掩蔽的状态开始,逐步预测并解除部分序列和侧链 token 的掩码,直至得到完整的序列和侧链结构。如下图所示:
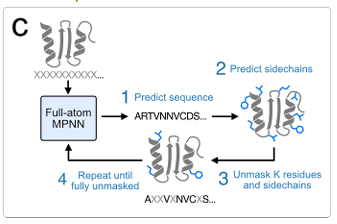
迭代采样策略
在具体设计上,侧链坐标表示采用 atom37 格式,每个残基均为 37 x 3 固定大小的矩阵,37 个原子包含 4 个主链原子(N、Cα、C 和 O)和 33 个侧链原子的三维坐标。对于不存在特定原子类型的侧链,使用幽灵原子(设为残基的 Cα 位置)表示。该方法解决了不同氨基酸侧链原子数量不同的问题。
作为特征提取核心,全原子编码器则使用了混合 MPNN-GVP 架构的图神经网络进行编码,该架构由 3 个主要组件构成:不变主链编码器、不变全原子编码器和等变全原子编码器。其中,前两个组件基于 ProteinMPNN 的架构构建,不变主链编码器与 MPNN 编码器相同,仅对主链结构进行编码;不变全原子编码器则取代 MPNN 解码器,其与主链编码器 MPNN 编码器相同,但特征化扩展到了所有原子。最后一个组件使用了改进的 GVP,是为了让模型除能够对先前编码的标量值原子间距离进行推理外,还能对向量值原子间取向进行推理。
在侧链坐标生成方面,研究团队采用了逐 token 欧几里得扩散(Per-token Euclidean Diffusion)方法。其核心是采用欧几里得扩散模型(EDM)解决侧链原子坐标这一连续值的生成问题,目标是生成与主链结构、周围氨基酸空间排布相匹配的侧链结构。在训练时,先给真实侧链坐标加随机噪声,再让模型根据噪声水平和已知信息去除噪声、恢复真实坐标;推理时,从随机噪声坐标开始,模型逐步去噪生成接近真实的侧链坐标。
同时,为了避免蛋白质整体旋转、平移对侧链生成的影响,训练时将侧链原子坐标转换到基于主链原子的局部坐标系,生成后再转回全局坐标系。该扩散模型的输入包含全原子编码器提取的特征、已预测的序列身份和当前噪声水平,生成的侧链坐标还会用于联合损失函数指导模型训练。如下图所示。
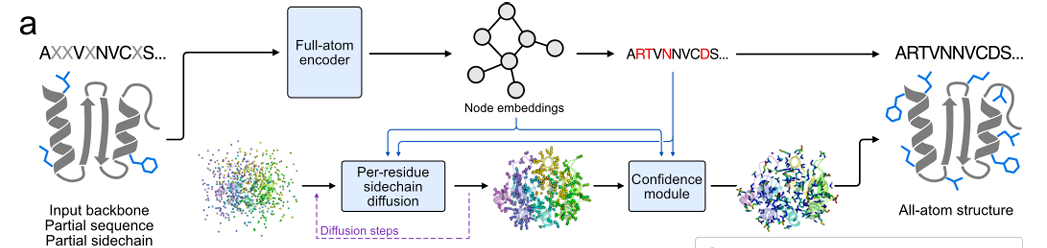
全原子序列设计概述
为了减少模型预测的误差,提升准确性,研究团队还设计了预测侧链 Packing 误差的置信度模块(Predicted Sidechain Error,pSCE)。具体来说,该模块通过将侧链原子的实际误差(生成坐标与真实坐标的距离)划分为 33 个区间,用分类交叉熵损失训练模型,使其能根据生成过程中的信息预测每个原子误差所在的区间,再通过区间概率的期望得到最终的误差估计值 pSCE。这一模块的输入包括全原子编码器的特征、生成的序列和侧链坐标以及扩散过程中的噪声水平,输出的 pSCE 能有效反映侧链 Packing 的精度,既有助于筛选高质量的设计结果,也可以增强模型的可解释性,从而完善了侧链结构生成的质量评估环节。
实验结果:性能显著优于仅基于主链的模型
为对模型进行性能验证和准确评估,研究团队首先进行了序列恢复和自洽性评估实验,并将 FAMPNN 与其他方法进行基准比较,具体比较对象如下图中所示。
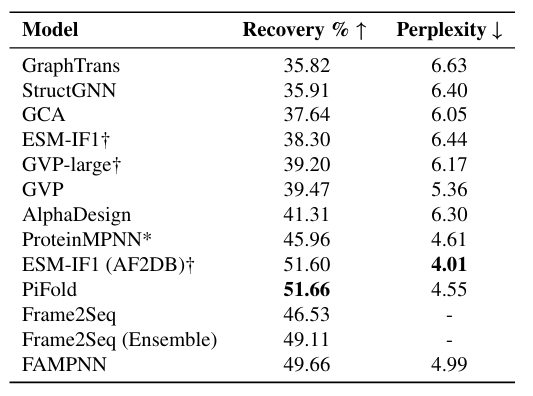
在 CATH 4.2 测试集上的中位序列准确率比较
实验显示,FAMPNN 在单步序列恢复准确率方面的表现超过了当前先进方法,达到了49.66%,作为对比,ProteinMPNN 仅为 45.96%,GVP 仅为 39.47%。
在基于 RFdiffusion 生成新主链上的自洽性评估中,FAMPNN(0.3Å)在 scTM (结构相似性)和 scRMSD (均方根偏差)指标上与 ProteinMPNN 相当,且 10 步迭代采样即可达到高度自洽,比全自回归方法更高效。如下图所示:
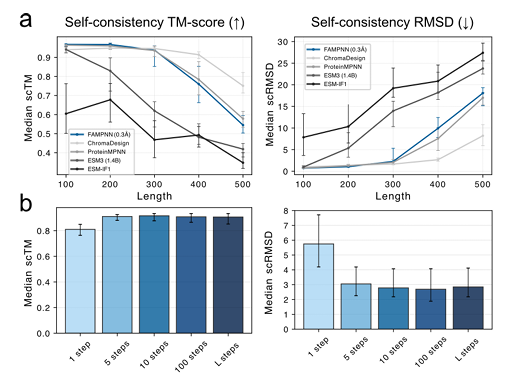
自洽性评估实验
在侧链 Packing 方面,研究人员在 CASP13、14、15 数据集上,将所提模型与其他方法进行了比较。实验显示,在 CASP15 测试集的晶体结构评估中,FAMPNN(0.0Å)的原子 RMSD (All / Core / Surface)为 0.690/0.350/0.789Å,优于其他方法,并且其与每个原子的误差和每个残基的误差都具有很强的相关性,斯皮尔曼相关系数(Spearman)分别为 0.843 和 0.780。如下图所示:
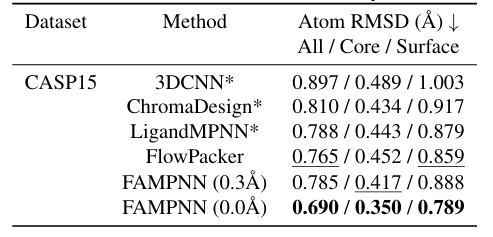
在 CASP 数据集上的侧链 packing 性能比较
在全原子条件下的蛋白质适应性评估中,在 SKEMPlv2 数据集上,FAMPNN 的表现显著优于无监督模型,甚至在测试子集上超过了监督模型,其在零样本预测中展示了强大的泛化能力。在 S669、Megascale 和 FireProtDB 三个稳定性数据集上,FAMPNN 的表现略优于 ProteinMPNN 和 ESM-IF;在抗体-抗原结合亲和力预测中,FAMPNN 始终优于最先进的无监督方法 ProteinMPNN 和 ESM-IF1,这证明了 FAMPNN 在蛋白质稳定性和增强蛋白质-蛋白质相互作用方面的效用。如下图所示:
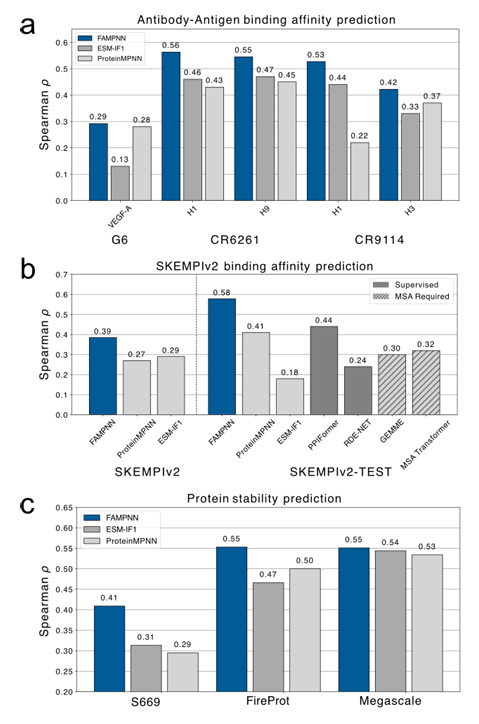
全原子条件下的蛋白质适应度评估,FAMPNN 与最先进蛋白质设计方法的性能比较
在评估全原子建模是否能够提高序列设计性能的实验中,研究发现添加侧链 Packing 目标和全原子条件设定,均可以提高序列准确性,并且 FAMPNN 和基线模型的性能都会随着更多结构信息注入而提升。在蛋白质-蛋白质界面处,侧链相互作用的建模更为关键,与仅提供部分序列上下文相比,结合序列提供部分侧链上下文可以明显提高准确性。
此外,与 LigandMPNN 相比,FAMPNN 能够更有效地利用侧链上下文,并且在基于不同数量的部分序列或侧链构象上下文的条件下进行侧链 Packing,上下文越多,包装准确率越高。如下图 c 所示:
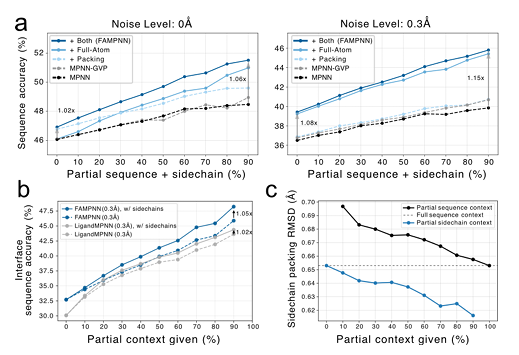
在 CATH 4.2 测试集上对模型训练、条件设定和架构的各种消融实验
总而言之,上述实验证明了 FAMPNN 在蛋白质适应性预测方面相较于仅基于主链的模型具有显著优势。
人工智能驱动,学术界在侧链建模领域百花齐放
正如开篇所提,侧链构象对蛋白质功能至关重要,而又因为蛋白质主链确定后,侧链构象仍存在多种可能,这使得侧链构象的建模和研究变成一件困难且必须攻克的问题。除本次研究之外,全球不少学术研究机构正通过前沿的深度学习技术和生物学知识,驱动侧链建模的研究向前发展。
中国的复旦大学团队提出了名为 OPUS-Rota5 的两阶段侧链建模方法。该方法利用改进的 3D-Unet 捕获局部环境特征,包括每个残基的配体信息,然后使用 RotaFormer 模块聚合各种类型特征。在包括 CAMEO、CASP15 等测试集上的评估表明,OPUS-Rota5 显著优于其他一些领先的侧链建模方法。相关研究以「OPUS-Rota5: A highly accurate protein side-chain modeling method with 3D-Unet and RotaFormer」为题,发布于 ScienceDirect。
论文地址:
https://www.sciencedirect.com/science/article/pii/S0969212624001266
来自北京大学的团队提出了另外一种方法,名为 GeoPacker,该方法结合几何深度学习与 ResNet 对蛋白质侧链进行建模。GeoPacker 通过具有旋转和平移不变性的方式明确表示原子相互作用,以提取相对位置信息。在侧链结构预测精度方面,GeoPacker 表现出优于基于能量函数的最先进方法,并且在预测精度相当的情况下,其运行速度分别比基于深度学习的方法 DLPacker 和 OPUS-Rota4 快了约 10 倍和 700 倍。相关研究以「GeoPacker: A novel deep learning framework for proteinside-chain modeling」发布。
论文地址:
https://onlinelibrary.wiley.com/doi/epdf/10.1002/pro.4484
同时,多伦多大学的团队提出了名为 FlowPacker 的模型,其目的是在已知蛋白质的氨基酸序列和主链结构的基础上,精准预测侧链的具体形态。与以往先进方法相比,FlowPacker 在大多数指标上表现更好,而且运行速度更快。比如在角度预测误差、预测角度与真实值的接近程度、原子位置偏差等方面都有优势。相关研究以「FlowPacker: Protein side-chain packing with torsional flow matching」为题发表。
论文地址:
https://www.mlsb.io/papers_2024/FlowPacker:_protein_side-chain_packing_with_torsional_flow_matching.pdf
总的来看,解码侧链构象对于生命科学领域的发展是至关重要的,而人工智能技术的不断发展,毫无疑问推动了结构生物学和计算生物学一路狂飙,也助力了国内外研究机构学术成果实现百花齐放,一旦这些成果从实验场落地到应用,势必将引起生命科学领域的新一轮风暴,推动生物科学、医药科学迈入全新的篇章。



戳“阅读原文”,免费获取海量数据集资源!
(文:HyperAI超神经)