
作者:梅菜
编辑:十九
转载请联系本公众号获得授权,并标明来源
来自剑桥大学的研究团队提出了一个名为 Celcomen 的虚拟组织模型,这是首个在空间转录组学分析中具有因果推断可识别性的模型,不仅能估计环境对单个细胞的影响,还能推测单个细胞对其周围环境及整体组织的影响。
在生物学领域,细胞的基因表达谱同时编码了其内在特性和外在组织微环境的信息。解开这两种效应的因果关系,对全面理解细胞内与细胞间的复杂相互作用十分关键。为此,需要一个稳健的因果解耦 (causal disentanglement) 框架。
因果解耦是一种机器学习方法,旨在通过揭示数据中的因果关系分离出有用的特征和无关的特征,从而减少模型对虚假关联的依赖,提高模型的鲁棒性和泛化能力。在因果解耦等机器学习理论发展的同时,生物学领域的技术进步也推动了空间转录组学的发展,使得研究人员可以在单细胞分辨率下同时测定细胞的基因表达和空间坐标,并能在空间样本中大规模开展基因敲除等扰动实验。
然而,当前处理空间转录组学的计算方法常常忽视了细胞和组织水平的因果扰动建模,而这一点对于揭示组织疾病状态背后的机制至关重要。例如,虚拟细胞 (Virtual Cells) 模型可以预测微环境和宏观环境 (如供体年龄、细胞所在组织、药物处理、gRNA 介导的基因敲除等) 变化对基因表达的影响,虚拟组织 (Virtual Tissues) 模型不仅能估计环境对单个细胞的影响,还能够推测单个细胞对其周围环境及整体组织的影响。
基于此,来自剑桥大学的研究团队提出了一个名为 Celcomen 的虚拟组织模型,其本质是一种基于数学因果关系的新型图神经网络,用于解开空间转录组学和单细胞数据中的细胞内和细胞间基因调控的秘密。研究人员验证了 Celcomen 在真实和自模拟的空间转录组学数据中解开并恢复基因-基因相互作用的能力。
相关成果以「Estimation of single-cell and tissue perturbation effect in spatial transcriptomics via Spatial Causal Disentanglement」为题,入选 ICLR 2025。
研究亮点:
* 研究证明了将虚拟细胞 (Virtual Cells) 模型推广至虚拟组织 (Virtual Tissues) 模型的可行性
* 研究提出了首个在空间转录组学分析中具有因果推断可识别性 (causally identifiable) 的模型
* 通过整合解离单细胞数据 (dissociated single-cell data) 和空间单细胞数据 (spatial single-cell data) 进行基因调控推断
论文地址:
https://openreview.net/forum?id=Tqdsruwyac
关注公众号,后台回复「Celcomen」获取完整 PDF
开源项目「awesome-ai4s」汇集了 200 余篇 AI4S 论文解读,并提供海量数据集与工具:
https://github.com/hyperai/awesome-ai4s
数据集:首次尝试利用 Perturbmap 数据集
为了展示 Celcomen 在空间背景下正确捕捉扰动效应的有效性,研究人员在体内全转录组数据集上进行了基准测试,该数据集测量了空间转录组学中的基因敲除,名为 Perturbmap。Perturbmap 数据集包含了一种小鼠模型,用于研究 KP 肺癌,此外,可能存在 Jak2 或 Tgfbr2 基因敲除。该数据集注释了 5 个空间区域作为病变区域,这些区域的部分是 1) KP 野生型癌症,或 2) KP 癌症与 Jak2 敲除,或 3) KP 癌症与 Tgfbr2 敲除,如下图所示:
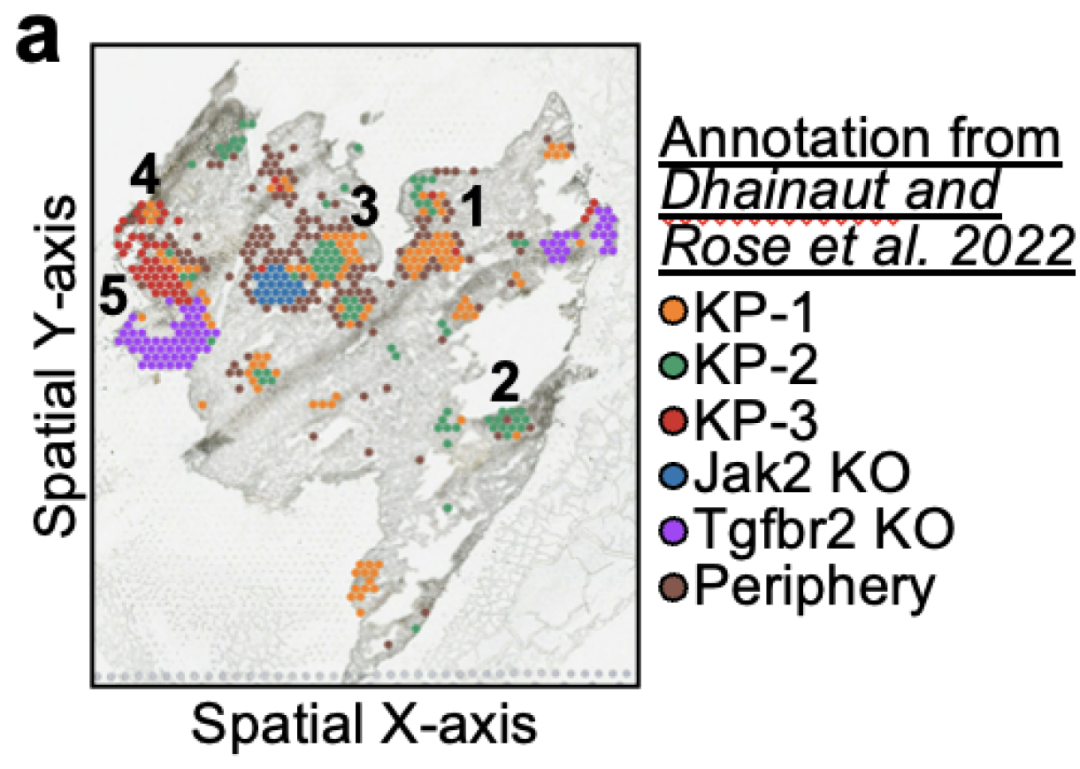
图:在空间轴上的散点图,每个 Visium 点的颜色表示肿瘤细胞表型
(KO 表示敲除,KP 表示野生型肿瘤)
在评估 Celcomen 能力的过程中,研究人员所使用的胎儿脾脏数据集来自 https://developmental.cellatlas.io/fetalimmune,以对数标准化形式提供,明确表示进行了对数变换和文库大小标准化;胶质母细胞瘤数据集来自 10x 基因组学,并进行了相同的文库大小标准化、百万计数 (CPM) 和对数变换,底数为 e;此外,仅保留了在至少 100 个细胞中表达的基因。
模型架构:一种新型因果分析框架 Celcomen
本研究提出的 Celcomen 模型通过结合拉格朗日力学 (Lagrangian mechanics) 和因果推断,实现了因果推断可识别性和更高的模型可解释性。可识别性简单来说,就是给定足够的数据和合理的假设,模型是否能明确地识别出因果关系,而不会因为多个不同的假设或模型设定而得出同样的观察结果——这为空间转录组学研究提供了一种新型的因果分析框架。
Celcomen 是基于 3 个核心假设构建的:①一阶邻居 (1st neighbors) 之间的基因-基因 (gene-gene) 期望相关性必须与观测数据完全匹配;②同一空间点/细胞内的基因-基因期望相关性必须与观测数据完全匹配;③因果充分性 (Causal Sufficiency) 假设:在研究的基因对之间,不存在未测量的共同原因。
如下图所示:Celcomen 分为推理模块 (CCE) 和生成模块 (SCE) 两部分:
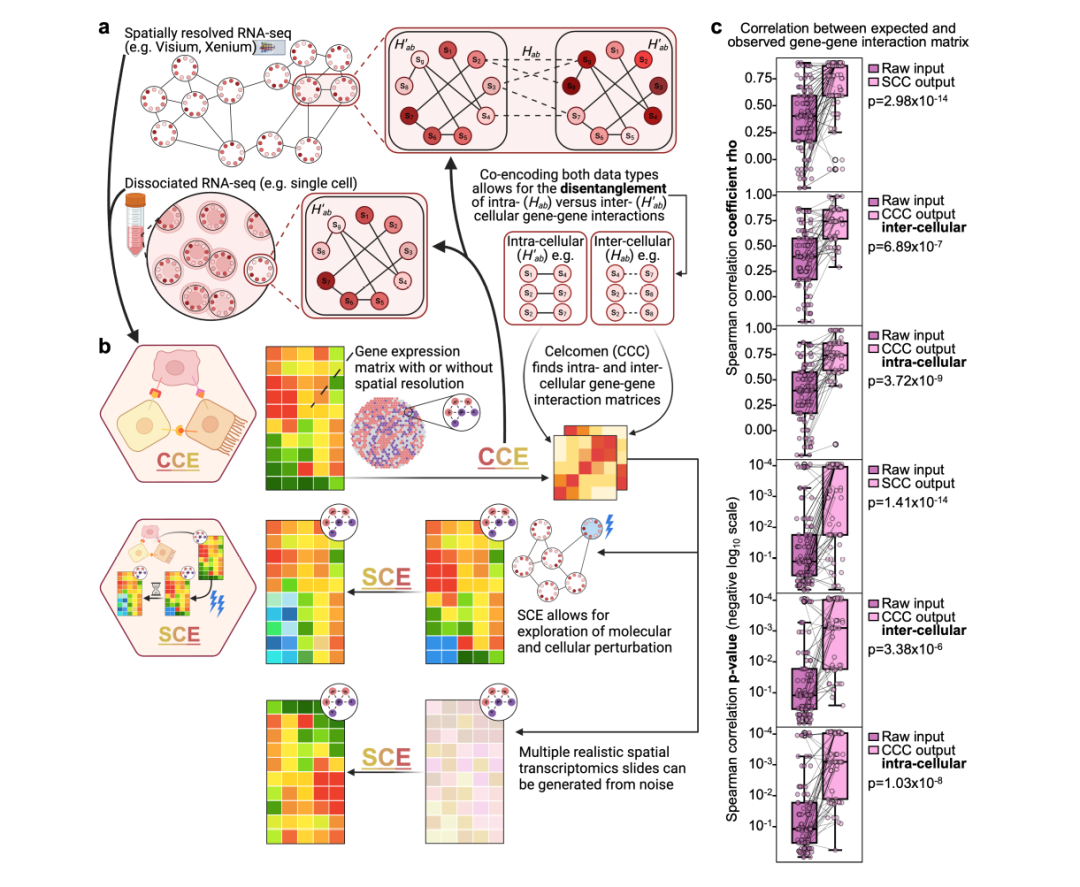
图:Celcomen 推理模块 (CCE) 和生成模块 (SCE)
(a) 推理模块 (CCE) :可以从空间解析转录组数据 (spatial RNA-seq data) 以及可选的解离单细胞 RNA-seq 数据 (dissociated scRNA-seq data) 中学习基因-基因 (gene-gene) 关系。在空间数据中,高亮的细胞-细胞对 (cell-cell pair),以及在单细胞 RNA-seq 数据中的单个细胞,展示了 CCE 如何区分细胞内 (H′ab)与细胞间 (Hab) 的基因-基因相互作用。
(b) 生成模块 (SCE):利用 CCE 学习到的基因-基因关系,以在细胞或基因扰动后模拟组织行为的反事实情境(counterfactual tissue behavior)。
* 反事实情境:这是一种用于研究生物组织在不同假设条件下可能呈现的行为的方法,主要用于因果推断、干预模拟和生物医学建模。它涉及构建一个假设的情境,即如果某个关键因素发生变化(如基因敲除、药物干预、外部环境变化等),生物组织的行为会如何不同于实际观测到的情况。
研究结果:Celcomen 模型在解开因果关系方面具有可识别性
研究人员通过自一致性合成数据和真实世界数据实验验证了 Celcomen 模型在因果结构学习和解开因果关系方面的可识别性。
Celcomen 具有强大的自一致性和可识别性
如下图所示,在合成数据集上,Celcomen 始终展示出其推断的基因-基因相互作用与真实数据之间的强烈一致性,表明 Celcomen 具有强大的自洽性 (self-consistency),因此也具有可识别性。
* 自洽性:在统计学、优化和机器学习中,自洽性通常意味着模型的假设、推导和优化过程能够收敛到一个稳定的解
* 可识别性:指的是在因果推断模型中,是否能够基于观察数据唯一确定因果关系的模型参数或因果效应
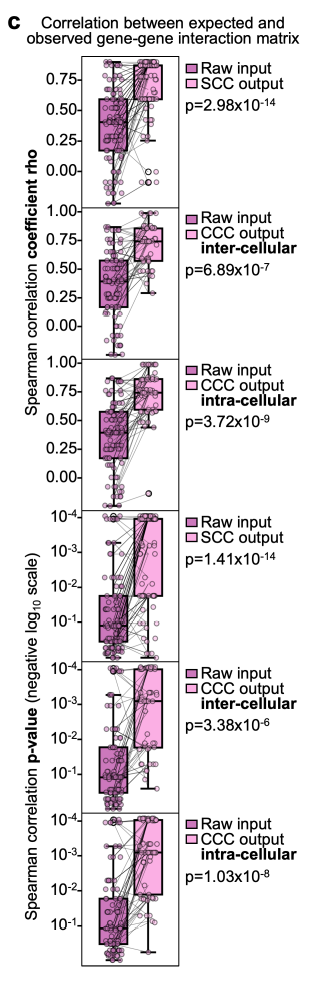
图:箱线图 (box plot)
x 轴表示用于比较的数据集
y 轴展示了斯皮尔曼相关系数(上方三图)和相关性 p 值(下方三图)
研究人员还通过将 Celcomen 模型应用于多个人类胎脾的空间转录组切片,以确认其在真实人类数据上的可识别性保证,结果观察到这两个基因-基因相互作用矩阵之间的斯皮尔曼相关系数在 0.5-0.6 范围内。此外,捕捉到的基因相互作用在细胞内和细胞间矩阵中是生物学上合理的,因为它们遵循已知的生物学细胞内和细胞间过程。
这证明了 Celcomen 的可识别性,确认了其隐含的稳定性和鲁棒性超越了理论和合成数据,且同样可以在真实人类样本中观察到。
因果解耦能力:Celcomen 能够成功解开内在与外在转录组变异的来源
接着,研究人员又测试了 Celcomen 解开细胞内与细胞间基因调控程序的能力 (解耦能力)。他们在一个真实的人类临床环境中应用 Celcomen,分析人类胶质母细胞瘤(大脑癌症)的单细胞分辨率空间转录组数据集,如下图所示,研究人员发现 Celcomen 能够成功地解开内在与外在转录组变异的来源。

图:Celcomen 在人类胶质母细胞瘤中再现已知的干扰素敲除生物学,并解析细胞内和细胞间基因-基因相互作用
体内空间反事实验证:与随机基准相比,Celcomen 的表现显著更高
为了进一步展示 Celcomen 的有效性,研究人员在体内全转录组数据集 Perturbmap 上进行了基准测试。结果显示,对于所有病变,预测与体内测量的斯皮尔曼相关性范围为 0.28-0.47。为了评估此性能的显著性,研究人员将模型与随机基准进行比较,其中 Celcomen 在随机打乱的数据上运行。结果发现,与随机基准相比,Celcomen 的表现显著更高,p 值为 0.0079,如下图 (c-f) 所示:
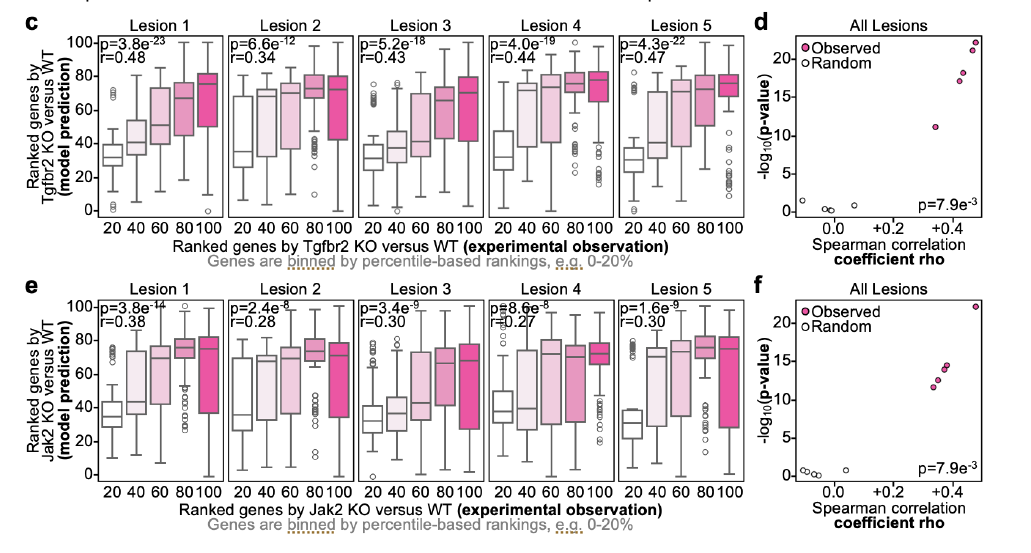
图:在肺癌模型中体内验证的反事实预测
c,e) 每个病变的箱型图
d,f) 散点图,每个点表示一个特定肿瘤病变区域
总而言之,研究提出的模型为通过因果推断实现机制可解释性开辟了新的道路。正如在实验中展示的,由于 Celcomen 模型的因果可辨识性,研究人员可以高精度地恢复神经网络的参数值。Celcomen 的进展显著影响生物医学领域,比如通过揭示疾病如何导致组织功能衰竭,促进有关治疗效益的可验证假设。随着技术的进步,Celcomen 的价值将不断增长,推动疾病建模和机制理解的提升。
人工智能充分释放空间转录组学潜力
本研究取得的相关成果是对空间转录组学的又一次发展——空间转录组技术是生物信息学领域近年来的重大突破之一,该技术通过提供详细的、空间定位的分子特征,极大地改变了生物医学研究范式,使生物学研究人员能够以前所未有的分辨率阐明组织结构和功能。
过去几年,空间转录组技术实现快速发展,数据得到不断的积累。在此基础上,2024 年 8 月发布的《Nature Methods 特刊评论:用人工智能之「钥」,开空间组学之「锁」》一文指出,人工智能有可能充分释放空间组学的潜力,促进复杂数据集的整合并发现新的生物医学结论。
具体而言,AI 可以促进空间转录组学与 scRNA-seq 的整合,使得研究人员在单细胞水平上测量转录组范围内的空间基因表达谱。此外,通过整合空间组学与组织学成像数据,AI 可以构建高分辨率、全面的三维空间组织图谱,覆盖广泛的组学模态。随着可用数据集数量的增长,多模态大语言模型 (MM-LLMs) 可以在空间组学、医学成像和临床文本数据上进行训练,用于生物医学研究和精准医学中的任务。
2023 年 10 月,中国科学院数学与系统科学研究院张世华课题组在 Nature Computational Science 上,发表了题为「Integrating spatial transcriptomics data across different conditions, technologies, and developmental stages」的研究论文。该工作针对来自不同技术、不同发育时间点、不同疾病条件的生物组织多切片空间转录组数据建立了整合分析新工具 STAligner,能帮助研究人员在进行空间转录组学分析时发现新的重要生物学见解。
*论文原文:
https://www.biorxiv.org/content/10.1101/2022.12.26.521888v1.full.pdf
为了解决空间转录组数据分析面临的多方面难题,2024 年 7 月,清华大学生命科学学院/结构生物学高精尖创新中心/清华-北大生命科学联合中心张强锋副教授课题组,在 Cell Systems 杂志在线发表题为「Tissue module discovery in single-cell resolution spatial transcriptomics data via cell-cell interaction-aware cell embedding」的研究论文。该研究开发了基于图自编码器 (Graph autoencoder) 深度学习框架的人工智能算法 SPACE (spatial transcriptomics data analysis via 「interaction-aware」 cell embedding),能够从单细胞分辨率的空间转录组数据中识别空间细胞类型和发现组织模块,可被用于大规模的空间转录组研究。
点击阅读更多详情:登 Cell 子刊!清华大学张强锋课题组开发 SPACE 算法,组织模块发现能力领先同类工具
展望未来,通过利用 AI 的强大计算能力和深度学习算法,研究人员有望解锁空间转录组学的全新维度,显著提升疾病研究、药物开发和个性化医学的效率,使科学家能够以前所未有的精度探索生物系统的空间异质性,从而带来开创性的科学发现。
参考资料:
1.https://openreview.net/forum?id=Tqdsruwyac
2.https://www.thepaper.cn/newsDetail_forward_28521641
3.https://www.cas.cn/syky/202310/t20231020_4981872.shtml
4.https://mp.weixin.qq.com/s/JnLixH6SBoLb0BIZTuF41w




戳“阅读原文”,免费获取海量数据集资源!
(文:HyperAI超神经)